
Running Jobs
There is one method for submitting jobs to Negishi. You may use SLURM to submit jobs to a partition on Negishi. SLURM performs job scheduling. Jobs may be any type of program. You may use either the batch or interactive mode to run your jobs. Use the batch mode for finished programs; use the interactive mode only for debugging.
In this section, you'll find a few pages describing the basics of creating and submitting SLURM jobs. As well, a number of example SLURM jobs that you may be able to adapt to your own needs.
Basics of SLURM Jobs
The Simple Linux Utility for Resource Management (SLURM) is a system providing job scheduling and job management on compute clusters. With SLURM, a user requests resources and submits a job to a queue. The system will then take jobs from queues, allocate the necessary nodes, and execute them.
Do NOT run large, long, multi-threaded, parallel, or CPU-intensive jobs on a front-end login host. All users share the front-end hosts, and running anything but the smallest test job will negatively impact everyone's ability to use Negishi. Always use SLURM to submit your work as a job.
Link to section 'Submitting a Job' of 'Basics of SLURM Jobs' Submitting a Job
The main steps to submitting a job are:
Follow the links below for information on these steps, and other basic information about jobs. A number of example SLURM jobs are also available.
Queues
On Negishi, the required options for job submission deviates from some of the other community clusters you might have experience using. In general every job submission will have four parts: “sbatch --ntasks=1 --cpus-per-task=4 --partition=cpu --account=rcac --qos=standby”
-
The number and type of resources you want (
--ntasks=1 --cpus-per-task=4) -
The partition where the resources are located (
--partition=cpu) -
The account the resources should come out of (
--account=rcac) -
The quality of service (QOS) this job expects from the resources (
--qos=standby)
| Use Case | Old Syntax | New Syntax |
|---|---|---|
| Submit a job to your group's account | sbatch -A mygroup |
sbatch -A mygroup -p cpu |
| Submit a standby job | sbatch -A standby |
sbatch -A mygroup -p cpu -q standby |
| Submit a highmem job | sbatch -A highmem |
sbatch -A mygroup -p highmem |
| Submit a gpu job | sbatch -A gpu |
sbatch -A mygroup -p gpu |
| Submit a job to your group's interactive account | sbatch -A interactive |
sbatch -A mygroup -p negishi -q interactive |
If you have used other clusters, you will be familiar with the first item. If you have not, you can read about how to format the request on our job submission page. The rest of this page will focus on the last three items.
Link to section 'Partitions' of 'Queues' Partitions
On Negishi, the various types of nodes on the cluster are organized into distinct partitions. This allows jobs to different node types to be charged separately and differently. This also means that Instead of only needing to specify the account name in the job script, the desired partition must also be specified. Each of these partitions is subject to different limitations and has a specific use case that will be described below.
Link to section 'CPU Partition' of 'Queues' CPU Partition
This partition contains the resources a group purchases access to when they purchase CPU resources on Negishi and is made up of 446 Bell-A nodes. Each of these nodes contains two Zen 3 AMD EPYC 7763 64-core processors for a total of 128 cores and 256 GB of memory for a total of more than 57,000 cores in the partition. Memory in this partition is allocated proportional to your core request such that each core is given about 2 GB of memory per core requested. Submission to this partition can be accomplished by using the option: -p cpu or --partition=cpu.
The purchasing model for this partition allows groups to purchase high priority access to some number of cores. When an account uses resources in this account by submitting a job tagged with the normal QOS, the cores used by that job are withdrawn from the account and deposited back into the account when the job terminates.
When using the CPU partition, jobs are tagged by the normal QOS by default, but they can be tagged with the standby QOS if explicitly submitted using the -q standby or --qos=standby option.
- Jobs tagged with the
normalQOS are subject to the following policies:- Jobs have a high priority and should not need to wait very long before starting.
- Any cores requested by these jobs are withdrawn from the account until the job terminates.
- These jobs can run for up to two weeks at a time.
- Jobs tagged with the
standbyQOS are subject to the following policies:- Jobs have a low priority and there is no expectation of job start time. If the partition is very busy with jobs using the
normalQOS or if you are requesting a very large job, then jobs using thestandbyQOS may take hours or days to start. - These jobs can use idle resources on the cluster and as such cores requested by these jobs are not withdrawn from the account to which they were submitted.
- These jobs can run for up to four hours at a time.
- Jobs have a low priority and there is no expectation of job start time. If the partition is very busy with jobs using the
Available QOSes: normal, standby
Link to section 'Highmem Partition' of 'Queues' Highmem Partition
This partition is made up of 6 Bell-B nodes which have four times as much memory as a standard Bell-A node, and access to this partition is given to all accounts on the cluster to enable work that has higher memory requirements. Each of these nodes contains two Zen 2 AMD EPYC 7763 64-core processors for a total of 128 cores and 1 TB of memory. Memory in this partition is allocated proportional to your core request such that each core is given about 8 GB of memory per core requested. Submission to this partition can be accomplished by using the option: -p highmem or --partition=highmem.
When using the Highmem partition, jobs are tagged by the normal QOS by default, and this is the only QOS that is available for this partition, so there is no need to specify a QOS when using this partition. Additionally jobs are tagged by a highmem partition QOS that enforces the following policies
- There is no expectation of job start time as these nodes are a shared resources that are given as a bonus for purchasing access to high priority access to resources on Negishi
- You can have 2 jobs running in this partition at once
- You can have 8 jobs submitted to thie partition at once
- Your jobs must use more than 64 of the 128 cores on the node otherwise your memory footprint would fit on a standard Negishi-A node
- These jobs can run for up to 24 hours at a time.
Available QOSes: normal
Link to section 'GPU Partition' of 'Queues' GPU Partition
This partition is made up of 5 Negishi-G nodes. Each of these nodes contains two AMD MI210s and two Zen 2 AMD EPYC 7313 16-core processors for a total of 32 cores and 512GB of memory. Memory in this partition is allocated proportional to your core request such that each core is given about 8 GB of memory per core requested. You should request cores proportional to the number of GPUs you are using in this partition (i.e. if you only need one of the two GPUs, you should request half of the cores on the node) Submission to this partition can be accomplished by using the option: -p gpu or --partition=gpu.
When using the gpu partition, jobs are tagged by the normal QOS by default, and this is the only QOS that is available for this partition, so there is no need to specify a QOS when using this partition. Additionally jobs are tagged by a gpu partition QOS that enforces the following policies
- There is no expectation of job start time as these nodes are a shared resources that are given as a bonus for purchasing access to high priority access to resources on Negishi
- You can use up to 2 GPUs in this partition at once
- You can have 8 jobs submitted to thie partition at once
- These jobs can run for up to 24 hours at a time.
Available QOSes: normal
Link to section 'Login Partition' of 'Queues' Login Partition
This partition contains the resources a group purchases access to when they purchase "interactive access" on Negishi. Interactive access allows submission of jobs directly to the front ends for immediate job start times. These jobs can only request up to 4 CPUs and 8 GB of memory each and interactive users can only have one job at a time. Submission to this partition can be accomplished by using the option: -p login or --partition=login. In order to use this partition, you must submit using the interactive QOS which enforces the following policies:
- You can have one job running at a time.
- You can use up to 4 cores and 8 GB of memory at a time.
- Jobs can run for up to 24 hours.
Available QOSes: interactive
Link to section 'Accounts' of 'Queues' Accounts
On the Negishi community cluster, users will have access to one or more accounts, also known as queues. These accounts are dedicated to and named after each partner who has purchased access to the cluster, and they provide partners and their researchers with priority access to their portion of the cluster. These accounts can be thought of as bank accounts that contain the resources a group has purchased access to which may include some number of cores. To see the list of accounts that you have access to on Negishi as well as the resources they contain, you can use the command slist.
On Negishi, you must explicitly define the account that you want to submit to using the -Aor--account= option.
Link to section 'Quality of Service (QOS)' of 'Queues' Quality of Service (QOS)
On Negishi, we use a Slurm concept called a Quality of Service or a QOS. A QOS can be thought of as a tag for a job that tells the scheduler how that job should be treated with respect to limits, priority, etc. The cluster administrators define the available QOSes as well as the policies for how each QOS should be treated on the cluster. A toy example of such a policy may be "no single user can have more than 200 jobs that has been tagged with a QOS named highpriority".
There are two classes of QOSes and a job can have both:
- Partition QOSes: A partition QOS is a tag that is automatically added to your job when you submit to a partition that defines a partition QOS.
- Job QOSes: A Job QOS is a tag that you explicitly give to a job using the option
-qor--qos=. By explicitly tagging your jobs this way, you can choose the policy that each one of your jobs should abide by. We will describe the policies for the available job QOSes in the partition section below.
As an extended metaphor, if we think of a job as a package that we need to have shipped to some destination, then the partition can be thought of as the carrier we decide to ship our package with. That carrier is going to have some company policies that dictate how you need to label/pack that package, and that company policy is like the partition QOS. It is the policy that is enforced for simply deciding to use that carrier, or in this case, deciding to submit to a particular partition.
The Job QOS can then be thought of as the various different types of shipping options that carrier might offer. You might pay extra to have that package shipped overnight. On the other hand you may choose to pay less and have your package arrive as available. Once we decide to go with a particular carrier, we are subject to their company policy, but we also have some degree of control through choosing one of their available shipping options. In the same way, when you choose to submit to a partition, you are subject to the limits enforced by the partition QOS, but you may be able to ask for your job to be handled a particular way by specifying a job QOS offered by the partition.
In order for a job to use a Job QOS, the user submitting the job must have access to the QOS, the account the job is being submitted to must accept the QOS, and the partition the job is being submitted to must accept the QOS. The below list of job QOSes are QOSes that every user and every account of Negishi has access to:
normal: ThenormalQOS is the default job QOS on the cluster meaning if you do not explicitly list an alternative job QOS, your job will be tagged with this QOS. The policy for this QOS provides a high priority and does not add any additional limits.standby: ThestandbyQOS must be explicitly used if desired by using the option-q standbyor--qos=standby. The policy for this QOS gives access to idle resources on the cluster. Jobs tagged with this QOS are "low priority" jobs and are only allowed to run for up to four hours at a time, however the resources used by these jobs do not count against the resources in your Account. For users of our previous clusters, usage of this QOS replaces the previous-A standbystyle of submission.
Some of these QOSes may not be available in every partition. Each of the partitions in the following section will enumerate which of these QOSes are allowed in the partition.
Job Submission Matrix
| Job Type | Partition | QoS | Job Submission Options | Number of Cores Per Account | Number of Jobs Per Account | Priority Accrual | Max Walltime |
|---|---|---|---|---|---|---|---|
| PI Queue | cpu | normal | -A "mygroup" -p cpu |
Limited to purchased cores | No limit | No Limit | 2 weeks |
| Standby Job | cpu | standby | -A "mygroup" -p cpu -q standby |
14272 Cores | 5000 | No Limit | 4 hours |
| Highmem Job | highmem | normal | -A "mygroup" -p highmem |
128 Cores | 2 | 1 | 24 hours |
| GPU Job | gpu | normal | -A "mygroup" -p gpu |
64 cores/3 GPUs/1 Node | 2 | 1 |
24 hours |
| Interactive-Tier Queues | interactive | normal | -A "mygroup" -p interactive |
4 cores | 1 per user | 1 | 24 hours |
Note: The normal QOS is the default and does not need to be specified.
Job Submission Script
To submit work to a SLURM queue, you must first create a job submission file. This job submission file is essentially a simple shell script. It will set any required environment variables, load any necessary modules, create or modify files and directories, and run any applications that you need:
#!/bin/bash
# FILENAME: myjobsubmissionfile
# Loads Matlab and sets the application up
module load matlab
# Change to the directory from which you originally submitted this job.
cd $SLURM_SUBMIT_DIR
# Runs a Matlab script named 'myscript'
matlab -nodisplay -singleCompThread -r myscript
Once your script is prepared, you are ready to submit your job.
Link to section 'Job Script Environment Variables' of 'Job Submission Script' Job Script Environment Variables
| Name | Description |
|---|---|
| SLURM_SUBMIT_DIR | Absolute path of the current working directory when you submitted this job |
| SLURM_JOBID | Job ID number assigned to this job by the batch system |
| SLURM_JOB_NAME | Job name supplied by the user |
| SLURM_JOB_NODELIST | Names of nodes assigned to this job |
| SLURM_CLUSTER_NAME | Name of the cluster executing the job |
| SLURM_SUBMIT_HOST | Hostname of the system where you submitted this job |
| SLURM_JOB_PARTITION | Name of the original queue to which you submitted this job |
Submitting a Job
Once you have a job submission file, you may submit this script to SLURM using the sbatch command. SLURM will find, or wait for, available resources matching your request and run your job there.
On Negishi, in order to submit jobs, you need to specify the partition, account and Quality of Service (QoS) name to which you want to submit your jobs. To familiarize yourself with the partitions and QoS available on Negishi, visit Negishi Queues and Partitions. To check the available partitions on Negishi, you can use the showpartitions , and to check your available accounts you can use slist commands. Slurm uses the term "Account" with the option -A or --account= to specify different batch accounts, the option -p or --partition= to select a specific partition for job submission, and the option -q or --qos= .
Partition statistics for cluster negishi at Wed Jul 30 13:07:17 EDT 2025
Partition #Nodes #CPU_cores Cores_pending Job_Nodes MaxJobTime Cores Mem/Node
Name State Total Idle Total Idle Resorc Other Min Max Day-hr:mn /node (GB)
negishi-nodes up$ 6 0 768 768 0 0 1 infin infinite 128 257
cpu up$ 446 0 57088 57067 0 163 1 infin infinite 128 257
highmem up$ 6 0 768 768 0 0 1 infin infinite 128 1031
cms up$ 16 0 4096 3840 0 9036 1 infin infinite 256 515
gpu up 5 0 160 0 0 30 1 infin infinite 32 515
negishi-login up$ 8 0 2048 2048 0 0 1 infin infinite 256 515
negishi-profiling up 4 3 512 512 0 0 1 infin infinite 128 257
Link to section 'CPU Partition' of 'Submitting a Job' CPU Partition
The CPU partition on Negishi has two Quality of Service (QoS) levels: normal and standby. To submit your job to one compute node on cpu partition and 'normal' QoS which has "high priority":
$ sbatch --nodes=1 --ntasks=1 --partition=cpu --account=accountname --qos=normal myjobsubmissionfile
$ sbatch -N1 -n1 -p cpu -A accountname -q normal myjobsubmissionfile
To submit your job to one compute node on cpu partition and 'standby' QoS which is has "low priority":
$ sbatch --nodes=1 --ntasks=1 --partition=cpu --account=accountname --qos=standby myjobsubmissionfile
$ sbatch -N1 -n1 -p cpu -A accountname -q standby myjobsubmissionfile
Link to section ' GPU Partition' of 'Submitting a Job' GPU Partition
On the GPU partition on Negishi you don’t need to specify the QoS name because only one QoS exists for this partition, and the default is normal. To submit your job to one compute node requesting one GPU on the gpu partition under the 'normal' QoS which has "high priority":
$ sbatch --nodes=1 --gpus-per-node=1 --ntasks=1 --cpus-per-task=64 --partition=gpu --account=accountname myjobsubmissionfile
$ sbatch -N1 --gpus-per-node=1 -n1 -c64 -p gpu -A accountname -q normal myjobsubmissionfile
Link to section 'Highmem Partition' of 'Submitting a Job' Highmem Partition
To submit your job to a compute node in the highmem partition, you don’t need to specify the QoS name because only one QoS exists for this partition, and the default is normal. However, the highmem partition is only suitable for jobs with memory requirements that exceed the capacity of a standard node, so the number of requested tasks should be appropriately high.
$ sbatch --nodes=1 --ntasks=1 --cpus-per-task=64 --partition=highmem --account=accountname myjobsubmissionfile
$ sbatch -N1 -n1 -c64 -p gpu -A accountname myjobsubmissionfile
Link to section 'Login Partition' of 'Submitting a Job' Login Partition
The login partition is reserved for Negishi users who have purchased "interactive" access to Negishi. As an access control, in order to submit to this partition, you must supply the interactive QoS as a job option as this is the only QoS accepted by the partition. This partition is meant to give near-immediate start times to computationally modest jobs.
$ sbatch --nodes=1 --ntasks=1 --cpus-per-task=4 --partition=login --qos=interactive --account=accountname myjobsubmissionfile
$ sbatch -N1 -n1 -c4 -p gpu -q interactive -A accountname myjobsubmissionfile
Link to section 'General Information' of 'Submitting a Job' General Information
By default, each job receives 30 minutes of wall time, or clock time. If you know that your job will not need more than a certain amount of time to run, request less than the maximum wall time, as this may allow your job to run sooner. To request 1 hour and 30 minutes of wall time:
$ sbatch -t 01:30:00 -N=1 -n=1 -p=cpu -A=accountname -q=standby myjobsubmissionfile
The --nodes= or -N value indicates how many compute nodes you would like for your job, and --ntasks= or -n value indicates the number of tasks you want to run.
In some cases, you may want to request multiple nodes. To utilize multiple nodes, you will need to have a program or code that is specifically programmed to use multiple nodes such as with MPI. Simply requesting more nodes will not make your work go faster. Your code must support this ability.
To request 2 compute nodes:
$ sbatch -t 01:30:00 -N=2 -n=16 -p=cpu -A=accountname -q=standby myjobsubmissionfile
By default, jobs on Negishi will share nodes with other jobs.
If more convenient, you may also specify any command line options to sbatch from within your job submission file, using a special form of comment:
#!/bin/sh -l
# FILENAME: myjobsubmissionfile
#SBATCH --account=accountname
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --partition=cpu
#SBATCH --qos=normal
#SBATCH --time=1:30:00
#SBATCH --job-name myjobname
# Print the hostname of the compute node on which this job is running.
/bin/hostname
If an option is present in both your job submission file and on the command line, the option on the command line will take precedence.
After you submit your job with SBATCH, it may wait in queue for minutes, hours, or even weeks. How long it takes for a job to start depends on the specific queue, the resources and time requested, and other jobs already waiting in that queue requested as well. It is impossible to say for sure when any given job will start. For best results, request no more resources than your job requires.
Once your job is submitted, you can monitor the job status, wait for the job to complete, and check the job output.
Checking Job Status
Once a job is submitted there are several commands you can use to monitor the progress of the job.
To see your jobs, use the squeue -u command and specify your username:
(Remember, in our SLURM environment a queue is referred to as an 'Account')
squeue -u myusername
JOBID ACCOUNT NAME USER ST TIME NODES NODELIST(REASON)
182792 standby job1 myusername R 20:19 1 a000
185841 standby job2 myusername R 20:19 1 a001
185844 standby job3 myusername R 20:18 1 a002
185847 standby job4 myusername R 20:18 1 a003
To retrieve useful information about your queued or running job, use the scontrol show job command with your job's ID number. The output should look similar to the following:
scontrol show job 3519
JobId=3519 JobName=t.sub
UserId=myusername GroupId=mygroup MCS_label=N/A
Priority=3 Nice=0 Account=(null) QOS=(null)
JobState=PENDING Reason=BeginTime Dependency=(null)
Requeue=1 Restarts=0 BatchFlag=1 Reboot=0 ExitCode=0:0
RunTime=00:00:00 TimeLimit=7-00:00:00 TimeMin=N/A
SubmitTime=2019-08-29T16:56:52 EligibleTime=2019-08-29T23:30:00
AccrueTime=Unknown
StartTime=2019-08-29T23:30:00 EndTime=2019-09-05T23:30:00 Deadline=N/A
PreemptTime=None SuspendTime=None SecsPreSuspend=0
LastSchedEval=2019-08-29T16:56:52
Partition=workq AllocNode:Sid=mack-fe00:54476
ReqNodeList=(null) ExcNodeList=(null)
NodeList=(null)
NumNodes=1 NumCPUs=2 NumTasks=2 CPUs/Task=1 ReqB:S:C:T=0:0:*:*
TRES=cpu=2,node=1,billing=2
Socks/Node=* NtasksPerN:B:S:C=0:0:*:* CoreSpec=*
MinCPUsNode=1 MinMemoryNode=0 MinTmpDiskNode=0
Features=(null) DelayBoot=00:00:00
OverSubscribe=OK Contiguous=0 Licenses=(null) Network=(null)
Command=/home/myusername/jobdir/myjobfile.sub
WorkDir=/home/myusername/jobdir
StdErr=/home/myusername/jobdir/slurm-3519.out
StdIn=/dev/null
StdOut=/home/myusername/jobdir/slurm-3519.out
Power=
There are several useful bits of information in this output.
JobStatelets you know if the job is Pending, Running, Completed, or Held.RunTime and TimeLimitwill show how long the job has run and its maximum time.SubmitTimeis when the job was submitted to the cluster.NumNodes,NumCPUs,NumTasksandCPUs/Taskare the number of Nodes, CPUs, Tasks, and CPUs per Task are shown.WorkDiris the job's working directory.StdOutandStderrare the locations of stdout and stderr of the job, respectively.Reasonwill show why aPENDINGjob isn't running. The above error says that it has been requested to start at a specific, later time.
Checking Job Output
Once a job is submitted, and has started, it will write its standard output and standard error to files that you can read.
SLURM catches output written to standard output and standard error - what would be printed to your screen if you ran your program interactively. Unless you specfied otherwise, SLURM will put the output in the directory where you submitted the job in a file named slurm- followed by the job id, with the extension out. For example slurm-3509.out. Note that both stdout and stderr will be written into the same file, unless you specify otherwise.
If your program writes its own output files, those files will be created as defined by the program. This may be in the directory where the program was run, or may be defined in a configuration or input file. You will need to check the documentation for your program for more details.
Link to section 'Redirecting Job Output' of 'Checking Job Output' Redirecting Job Output
It is possible to redirect job output to somewhere other than the default location with the --error and --output directives:
#!/bin/bash
#SBATCH --output=/home/myusername/joboutput/myjob.out
#SBATCH --error=/home/myusername/joboutput/myjob.out
# This job prints "Hello World" to output and exits
echo "Hello World"
Job Dependencies
Dependencies are an automated way of holding and releasing jobs. Jobs with a dependency are held until the condition is satisfied. Once the condition is satisfied jobs only then become eligible to run and must still queue as normal.
Job dependencies may be configured to ensure jobs start in a specified order. Jobs can be configured to run after other job state changes, such as when the job starts or the job ends.
These examples illustrate setting dependencies in several ways. Typically dependencies are set by capturing and using the job ID from the last job submitted.
To run a job after job myjobid has started:
sbatch --dependency=after:myjobid myjobsubmissionfile
To run a job after job myjobid ends without error:
sbatch --dependency=afterok:myjobid myjobsubmissionfile
To run a job after job myjobid ends with errors:
sbatch --dependency=afternotok:myjobid myjobsubmissionfile
To run a job after job myjobid ends with or without errors:
sbatch --dependency=afterany:myjobid myjobsubmissionfile
To set more complex dependencies on multiple jobs and conditions:
sbatch --dependency=after:myjobid1:myjobid2:myjobid3,afterok:myjobid4 myjobsubmissionfile
Holding a Job
Sometimes you may want to submit a job but not have it run just yet. You may be wanting to allow lab mates to cut in front of you in the queue - so hold the job until their jobs have started, and then release yours.
To place a hold on a job before it starts running, use the scontrol hold job command:
$ scontrol hold job myjobid
Once a job has started running it can not be placed on hold.
To release a hold on a job, use the scontrol release job command:
$ scontrol release job myjobid
You find the job ID using the squeue command as explained in the SLURM Job Status section.
Canceling a Job
To stop a job before it finishes or remove it from a queue, use the scancel command:
scancel myjobid
You find the job ID using the squeue command as explained in the SLURM Job Status section.
Example Jobs
A number of example jobs are available for you to look over and adapt to your own needs. The first few are generic examples, and latter ones go into specifics for particular software packages.
Generic SLURM Jobs
The following examples demonstrate the basics of SLURM jobs, and are designed to cover common job request scenarios. These example jobs will need to be modified to run your application or code.
Simple Job
Every SLURM job consists of a job submission file. A job submission file contains a list of commands that run your program and a set of resource (nodes, walltime, queue) requests. The resource requests can appear in the job submission file or can be specified at submit-time as shown below.
This simple example submits the job submission file hello.sub to the standby queue on Negishi and requests a single node:
#!/bin/bash
# FILENAME: hello.sub
# Show this ran on a compute node by running the hostname command.
hostname
echo "Hello World"
sbatch -A standby --nodes=1 --ntasks=1 --cpus-per-task=1 --time=00:01:00 hello.sub
Submitted batch job 3521
For a real job you would replace echo "Hello World" with a command, or sequence of commands, that run your program.
After your job finishes running, the ls command will show a new file in your directory, the .out file:
ls -l
hello.sub
slurm-3521.out
The file slurm-3521.out contains the output and errors your program would have written to the screen if you had typed its commands at a command prompt:
cat slurm-3521.out
a001.negishi.rcac.purdue.edu
Hello World
You should see the hostname of the compute node your job was executed on. Following should be the "Hello World" statement.
Multiple Node
In some cases, you may want to request multiple nodes. To utilize multiple nodes, you will need to have a program or code that is specifically programmed to use multiple nodes such as with MPI. Simply requesting more nodes will not make your work go faster. Your code must support this ability.
This example shows a request for multiple compute nodes. The job submission file contains a single command to show the names of the compute nodes allocated:
# FILENAME: myjobsubmissionfile.sub
#!/bin/bash
echo "$SLURM_JOB_NODELIST"
sbatch --nodes=2 --ntasks=256 --time=00:10:00 -A standby myjobsubmissionfile.sub
Compute nodes allocated:
a[014-015].negishi
The above example will allocate the total of 256 CPU cores across 2 nodes. Note that if your multi-node job requests fewer than each node's full 128 cores per node, by default Slurm provides no guarantee with respect to how this total is distributed between assigned nodes (i.e. the cores may not necessarily be split evenly). If you need specific arrangements of your tasks and cores, you can use --cpus-per-task= and/or --ntasks-per-node= flags. See Slurm documentation or man sbatch for more options.
Directives
So far these examples have shown submitting jobs with the resource requests on the sbatch command line such as:
sbatch -A standby --nodes=1 --time=00:01:00 hello.sub
The resource requests can also be put into job submission file itself. Documenting the resource requests in the job submission is desirable because the job can be easily reproduced later. Details left in your command history are quickly lost. Arguments are specified with the #SBATCH syntax:
#!/bin/bash
# FILENAME: hello.sub
#SBATCH -A standby
#SBATCH --nodes=1 --time=00:01:00
# Show this ran on a compute node by running the hostname command.
hostname
echo "Hello World"
The #SBATCH directives must appear at the top of your submission file. SLURM will stop parsing directives as soon as it encounters a line that does not start with '#'. If you insert a directive in the middle of your script, it will be ignored.
This job can be then submitted with:
sbatch hello.sub
Specific Types of Nodes
SLURM allows running a job on specific types of compute nodes to accommodate special hardware requirements (e.g. a certain CPU or GPU type, etc.)
Cluster nodes have a set of descriptive features assigned to them, and users can specify which of these features are required by their job by using the constraint option at submission time. Only nodes having features matching the job constraints will be used to satisfy the request.
Example: a job requires a compute node in an "A" sub-cluster:
sbatch --nodes=1 --ntasks=128 --constraint=A myjobsubmissionfile.subCompute node allocated:
a003.negishiFeature constraints can be used for both batch and interactive jobs, as well as for individual job steps inside a job. Multiple constraints can be specified with a predefined syntax to achieve complex request logic (see detailed description of the '--constraint' option in man sbatch or online Slurm documentation).
Refer to Detailed Hardware Specification section for list of available sub-cluster labels, their respective per-node memory sizes and other hardware details. You could also use sfeatures command to list available constraint feature names for different node types.
Interactive Jobs
Interactive jobs are run on compute nodes, while giving you a shell to interact with. They give you the ability to type commands or use a graphical interface in the same way as if you were on a front-end login host.
To submit an interactive job, use sinteractive to run a login shell on allocated resources.
sinteractive accepts most of the same resource requests as sbatch, so to request a login shell on the cpu account while allocating 2 nodes and 128 total cores, you might do:
sinteractive -A cpu -N2 -n256
To quit your interactive job:
exit or Ctrl-D
The above example will allocate the total of 256 CPU cores across 2 nodes. Note that if your multi-node job requests fewer than each node's full 128 cores per node, by default Slurm provides no guarantee with respect to how this total is distributed between assigned nodes (i.e. the cores may not necessarily be split evenly). If you need specific arrangements of your tasks and cores, you can use --cpus-per-task= and/or --ntasks-per-node= flags. See Slurm documentation or man salloc for more options.
Serial Jobs
This shows how to submit one of the serial programs compiled in the section Compiling Serial Programs.
Create a job submission file:
#!/bin/bash
# FILENAME: serial_hello.sub
./serial_hello
Submit the job:
sbatch --nodes=1 --ntasks=1 --time=00:01:00 serial_hello.sub
After the job completes, view results in the output file:
cat slurm-myjobid.out
Runhost:a009.negishi.rcac.purdue.edu
hello, world
If the job failed to run, then view error messages in the file slurm-myjobid.out.
OpenMP
A shared-memory job is a single process that takes advantage of a multi-core processor and its shared memory to achieve parallelization.
This example shows how to submit an OpenMP program compiled in the section Compiling OpenMP Programs.
When running OpenMP programs, all threads must be on the same compute node to take advantage of shared memory. The threads cannot communicate between nodes.
To run an OpenMP program, set the environment variable OMP_NUM_THREADS to the desired number of threads:
In csh:
setenv OMP_NUM_THREADS 128In bash:
export OMP_NUM_THREADS=128This should almost always be equal to the number of cores on a compute node. You may want to set to another appropriate value if you are running several processes in parallel in a single job or node.
Create a job submissionfile:
#!/bin/bash
# FILENAME: omp_hello.sub
#SBATCH --nodes=1
#SBATCH --ntasks=128
#SBATCH --time=00:01:00
export OMP_NUM_THREADS=128
./omp_hello
Submit the job:
sbatch omp_hello.subView the results from one of the sample OpenMP programs about task parallelism:
cat omp_hello.sub.omyjobid
SERIAL REGION: Runhost:a003.negishi.rcac.purdue.edu Thread:0 of 1 thread hello, world
PARALLEL REGION: Runhost:a003.negishi.rcac.purdue.edu Thread:0 of 128 threads hello, world
PARALLEL REGION: Runhost:a003.negishi.rcac.purdue.edu Thread:1 of 128 threads hello, world
...If the job failed to run, then view error messages in the file slurm-myjobid.out.
If an OpenMP program uses a lot of memory and 128 threads use all of the memory of the compute node, use fewer processor cores (OpenMP threads) on that compute node.
MPI
An MPI job is a set of processes that take advantage of multiple compute nodes by communicating with each other. OpenMPI and Intel MPI (IMPI) are implementations of the MPI standard.
This section shows how to submit one of the MPI programs compiled in the section Compiling MPI Programs.
Use module load to set up the paths to access these libraries. Use module avail to see all MPI packages installed on Negishi.
Create a job submission file:
#!/bin/bash
# FILENAME: mpi_hello.sub
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=128
#SBATCH --time=00:01:00
#SBATCH -A standby
srun -n 256 ./mpi_hello
SLURM can run an MPI program with the srun command. The number of processes is requested with the -n option. If you do not specify the -n option, it will default to the total number of processor cores you request from SLURM.
If the code is built with OpenMPI, it can be run with a simple srun -n command. If it is built with Intel IMPI, then you also need to add the --mpi=pmi2 option: srun --mpi=pmi2 -n 256 ./mpi_hello in this example.
Submit the MPI job:
sbatch ./mpi_hello.subView results in the output file:
cat slurm-myjobid.out
Runhost:a010.negishi.rcac.purdue.edu Rank:0 of 256 ranks hello, world
Runhost:a010.negishi.rcac.purdue.edu Rank:1 of 256 ranks hello, world
...
Runhost:a011.negishi.rcac.purdue.edu Rank:128 of 256 ranks hello, world
Runhost:a011.negishi.rcac.purdue.edu Rank:129 of 256 ranks hello, world
...If the job failed to run, then view error messages in the output file.
If an MPI job uses a lot of memory and 128 MPI ranks per compute node use all of the memory of the compute nodes, request more compute nodes, while keeping the total number of MPI ranks unchanged.
Submit the job with double the number of compute nodes and modify the resource request to halve the number of MPI ranks per compute node.
#!/bin/bash
# FILENAME: mpi_hello.sub
#SBATCH --nodes=4
#SBATCH --ntasks-per-node=64
#SBATCH -t 00:01:00
#SBATCH -A standby
srun -n 256 ./mpi_hello
sbatch ./mpi_hello.subView results in the output file:
cat slurm-myjobid.out
Runhost:a010.negishi.rcac.purdue.edu Rank:0 of 256 ranks hello, world
Runhost:a010.negishi.rcac.purdue.edu Rank:1 of 256 ranks hello, world
...
Runhost:a011.negishi.rcac.purdue.edu Rank:64 of 256 ranks hello, world
...
Runhost:a012.negishi.rcac.purdue.edu Rank:128 of 256 ranks hello, world
...
Runhost:a013.negishi.rcac.purdue.edu Rank:192 of 256 ranks hello, world
...Notes
- Use slist to determine which queues (
--accountor-Aoption) are available to you. The name of the queue which is available to everyone on Negishi is "standby". - Invoking an MPI program on Negishi with ./program is typically wrong, since this will use only one MPI process and defeat the purpose of using MPI. Unless that is what you want (rarely the case), you should use srun or mpiexec to invoke an MPI program.
- In general, the exact order in which MPI ranks output similar write requests to an output file is random.
Link to section 'Collecting System Resource Utilization Data' of 'Monitoring Resources' Collecting System Resource Utilization Data
Knowing the precise resource utilization an application had during a job, such as CPU load or memory, can be incredibly useful. This is especially the case when the application isn't performing as expected.
One approach is to run a program like htop during an interactive job and keep an eye on system resources. You can get precise time-series data from nodes associated with your job using XDmod as well, online. But these methods don't gather telemetry in an automated fashion, nor do they give you control over the resolution or format of the data.
As a matter of course, a robust implementation of some HPC workload would include resource utilization data as a diagnostic tool in the event of some failure.
The monitor utility is a simple command line system resource monitoring tool for gathering such telemetry and is available as a module.
module load monitor
Complete documentation is available online at resource-monitor.readthedocs.io. A full manual page is also available for reference, man monitor.
In the context of a SLURM job you will need to put this monitoring task in the background to allow the rest of your job script to proceed. Be sure to interrupt these tasks at the end of your job.
#!/bin/bash
# FILENAME: monitored_job.sh
module load monitor
# track per-code CPU load
monitor cpu percent --all-cores >cpu-percent.log &
CPU_PID=$!
# track memory usage
monitor cpu memory >cpu-memory.log &
MEM_PID=$!
# your code here
# shut down the resource monitors
kill -s INT $CPU_PID $MEM_PID
A particularly elegant solution would be to include such tools in your prologue script and have the tear down in your epilogue script.
For large distributed jobs spread across multiple nodes, mpiexec can be used to gather telemetry from all nodes in the job. The hostname is included in each line of output so that data can be grouped as such. A concise way of constructing the needed list of hostnames in SLURM is to simply use srun hostname | sort -u.
#!/bin/bash
# FILENAME: monitored_job.sh
module load monitor
# track all CPUs (one monitor per host)
mpiexec -machinefile <(srun hostname | sort -u) \
monitor cpu percent --all-cores >cpu-percent.log &
CPU_PID=$!
# track memory on all hosts (one monitor per host)
mpiexec -machinefile <(srun hostname | sort -u) \
monitor cpu memory >cpu-memory.log &
MEM_PID=$!
# your code here
# shut down the resource monitors
kill -s INT $CPU_PID $MEM_PID
To get resource data in a more readily computable format, the monitor program can be told to output in CSV format with the --csv flag.
monitor cpu memory --csv >cpu-memory.csv
For a distributed job you will need to suppress the header lines otherwise one will be created by each host.
monitor cpu memory --csv | head -1 >cpu-memory.csv
mpiexec -machinefile <(srun hostname | sort -u) \
monitor cpu memory --csv --no-header >>cpu-memory.csv
Specific Applications
The following examples demonstrate job submission files for some common real-world applications. See the Generic SLURM Examples section for more examples on job submissions that can be adapted for use.
Gaussian
Gaussian is a computational chemistry software package which works on electronic structure. This section illustrates how to submit a small Gaussian job to a Slurm queue. This Gaussian example runs the Fletcher-Powell multivariable optimization.
Prepare a Gaussian input file with an appropriate filename, here named myjob.com. The final blank line is necessary:
#P TEST OPT=FP STO-3G OPTCYC=2
STO-3G FLETCHER-POWELL OPTIMIZATION OF WATER
0 1
O
H 1 R
H 1 R 2 A
R 0.96
A 104.
To submit this job, load Gaussian then run the provided script, named subg16. This job uses one compute node with 128 processor cores:
module load gaussian16
subg16 myjob -N 1 -n 128
View job status:
squeue -u myusername
View results in the file for Gaussian output, here named myjob.log. Only the first and last few lines appear here:
Entering Gaussian System, Link 0=/apps/cent7/gaussian/g16-A.03/g16-haswell/g16/g16
Initial command:
/apps/cent7/gaussian/g16-A.03/g16-haswell/g16/l1.exe /scratch/negishi/myusername/gaussian/Gau-7781.inp -scrdir=/scratch/negishi/myusername/gaussian/
Entering Link 1 = /apps/cent7/gaussian/g16-A.03/g16-haswell/g16/l1.exe PID= 7782.
Copyright (c) 1988,1990,1992,1993,1995,1998,2003,2009,2016,
Gaussian, Inc. All Rights Reserved.
.
.
.
Job cpu time: 0 days 0 hours 3 minutes 28.2 seconds.
Elapsed time: 0 days 0 hours 0 minutes 12.9 seconds.
File lengths (MBytes): RWF= 17 Int= 0 D2E= 0 Chk= 2 Scr= 2
Normal termination of Gaussian 16 at Tue May 1 17:12:00 2018.
real 13.85
user 202.05
sys 6.12
Machine:
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
a012.negishi.rcac.purdue.edu
Link to section 'Examples of Gaussian SLURM Job Submissions' of 'Gaussian' Examples of Gaussian SLURM Job Submissions
Submit job using 128 processor cores on a single node:
subg16 myjob -N 1 -n 128 -t 200:00:00 -A myqueuename
Submit job using 128 processor cores on each of 2 nodes:
subg16 myjob -N 2 --ntasks-per-node=128 -t 200:00:00 -A myqueuename
To submit a bash job, a submit script sample looks like:
#!/bin/bash
#SBATCH -A myqueuename # Queue name(use 'slist' command to find queues' name)
#SBATCH --nodes=1 # Total # of nodes
#SBATCH --ntasks=64 # Total # of MPI tasks
#SBATCH --time=1:00:00 # Total run time limit (hh:mm:ss)
#SBATCH -J myjobname # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
module load gaussian16
g16 < myjob.com
For more information about Gaussian:
Matlab
MATLAB® (MATrix LABoratory) is a high-level language and interactive environment for numerical computation, visualization, and programming. MATLAB is a product of MathWorks.
MATLAB, Simulink, Compiler, and several of the optional toolboxes are available to faculty, staff, and students. To see the kind and quantity of all MATLAB licenses plus the number that you are currently using you can use the matlab_licenses command:
$ module load matlab
$ matlab_licenses
The MATLAB client can be run in the front-end for application development, however, computationally intensive jobs must be run on compute nodes.
The following sections provide several examples illustrating how to submit MATLAB jobs to a Linux compute cluster.
Matlab Script (.m File)
This section illustrates how to submit a small, serial, MATLAB program as a job to a batch queue. This MATLAB program prints the name of the run host and gets three random numbers.
Prepare a MATLAB script myscript.m, and a MATLAB function file myfunction.m:
% FILENAME: myscript.m
% Display name of compute node which ran this job.
[c name] = system('hostname');
fprintf('\n\nhostname:%s\n', name);
% Display three random numbers.
A = rand(1,3);
fprintf('%f %f %f\n', A);
quit;
% FILENAME: myfunction.m
function result = myfunction ()
% Return name of compute node which ran this job.
[c name] = system('hostname');
result = sprintf('hostname:%s', name);
% Return three random numbers.
A = rand(1,3);
r = sprintf('%f %f %f', A);
result=strvcat(result,r);
end
Also, prepare a job submission file, here named myjob.sub. Run with the name of the script:
#!/bin/bash
# FILENAME: myjob.sub
echo "myjob.sub"
# Load module, and set up environment for Matlab to run
module load matlab
unset DISPLAY
# -nodisplay: run MATLAB in text mode; X11 server not needed
# -singleCompThread: turn off implicit parallelism
# -r: read MATLAB program; use MATLAB JIT Accelerator
# Run Matlab, with the above options and specifying our .m file
matlab -nodisplay -singleCompThread -r myscript
myjob.sub
< M A T L A B (R) >
Copyright 1984-2011 The MathWorks, Inc.
R2011b (7.13.0.564) 64-bit (glnxa64)
August 13, 2011
To get started, type one of these: helpwin, helpdesk, or demo.
For product information, visit www.mathworks.com.
hostname:a001.negishi.rcac.purdue.edu
0.814724 0.905792 0.126987
Output shows that a processor core on one compute node (a001) processed the job. Output also displays the three random numbers.
For more information about MATLAB:
Implicit Parallelism
MATLAB implements implicit parallelism which is automatic multithreading of many computations, such as matrix multiplication, linear algebra, and performing the same operation on a set of numbers. This is different from the explicit parallelism of the Parallel Computing Toolbox.
MATLAB offers implicit parallelism in the form of thread-parallel enabled functions. Since these processor cores, or threads, share a common memory, many MATLAB functions contain multithreading potential. Vector operations, the particular application or algorithm, and the amount of computation (array size) contribute to the determination of whether a function runs serially or with multithreading.
When your job triggers implicit parallelism, it attempts to allocate its threads on all processor cores of the compute node on which the MATLAB client is running, including processor cores running other jobs. This competition can degrade the performance of all jobs running on the node.
When you know that you are coding a serial job but are unsure whether you are using thread-parallel enabled operations, run MATLAB with implicit parallelism turned off. Beginning with the R2009b, you can turn multithreading off by starting MATLAB with -singleCompThread:
$ matlab -nodisplay -singleCompThread -r mymatlabprogram
When you are using implicit parallelism, make sure you request exclusive access to a compute node, as MATLAB has no facility for sharing nodes.
For more information about MATLAB's implicit parallelism:
Profile Manager
MATLAB offers two kinds of profiles for parallel execution: the 'local' profile and user-defined cluster profiles. The 'local' profile runs a MATLAB job on the processor core(s) of the same compute node, or front-end, that is running the client. To run a MATLAB job on compute node(s) different from the node running the client, you must define a Cluster Profile using the Cluster Profile Manager.
To prepare a user-defined cluster profile, use the Cluster Profile Manager in the Parallel menu. This profile contains the scheduler details (queue, nodes, processors, walltime, etc.) of your job submission. Ultimately, your cluster profile will be an argument to MATLAB functions like batch().
For your convenience, a generic cluster profile is provided that can be downloaded: myslurmprofile.settings
Please note that modifications are very likely to be required to make myslurmprofile.settings work. You may need to change values for number of nodes, number of workers, walltime, and submission queue specified in the file. As well, the generic profile itself depends on the particular job scheduler on the cluster, so you may need to download or create two or more generic profiles under different names. Each time you run a job using a Cluster Profile, make sure the specific profile you are using is appropriate for the job and the cluster.
To import the profile, start a MATLAB session and select Manage Cluster Profiles... from the Parallel menu. In the Cluster Profile Manager, select Import, navigate to the folder containing the profile, select myslurmprofile.settings and click OK. Remember that the profile will need to be customized for your specific needs. If you have any questions, please contact us.
For detailed information about MATLAB's Parallel Computing Toolbox, examples, demos, and tutorials:
Parallel Computing Toolbox (parfor)
The MATLAB Parallel Computing Toolbox (PCT) extends the MATLAB language with high-level, parallel-processing features such as parallel for loops, parallel regions, message passing, distributed arrays, and parallel numerical methods. It offers a shared-memory computing environment running on the local cluster profile in addition to your MATLAB client. Moreover, the MATLAB Distributed Computing Server (DCS) scales PCT applications up to the limit of your DCS licenses.
This section illustrates the fine-grained parallelism of a parallel for loop (parfor) in a pool job.
The following examples illustrate a method for submitting a small, parallel, MATLAB program with a parallel loop (parfor statement) as a job to a queue. This MATLAB program prints the name of the run host and shows the values of variables numlabs and labindex for each iteration of the parfor loop.
This method uses the job submission command to submit a MATLAB client which calls the MATLAB batch() function with a user-defined cluster profile.
Prepare a MATLAB pool program in a MATLAB script with an appropriate filename, here named myscript.m:
% FILENAME: myscript.m
% SERIAL REGION
[c name] = system('hostname');
fprintf('SERIAL REGION: hostname:%s\n', name)
numlabs = parpool('poolsize');
fprintf(' hostname numlabs labindex iteration\n')
fprintf(' ------------------------------- ------- -------- ---------\n')
tic;
% PARALLEL LOOP
parfor i = 1:8
[c name] = system('hostname');
name = name(1:length(name)-1);
fprintf('PARALLEL LOOP: %-31s %7d %8d %9d\n', name,numlabs,labindex,i)
pause(2);
end
% SERIAL REGION
elapsed_time = toc; % get elapsed time in parallel loop
fprintf('\n')
[c name] = system('hostname');
name = name(1:length(name)-1);
fprintf('SERIAL REGION: hostname:%s\n', name)
fprintf('Elapsed time in parallel loop: %f\n', elapsed_time)
The execution of a pool job starts with a worker executing the statements of the first serial region up to the parfor block, when it pauses. A set of workers (the pool) executes the parfor block. When they finish, the first worker resumes by executing the second serial region. The code displays the names of the compute nodes running the batch session and the worker pool.
Prepare a MATLAB script that calls MATLAB function batch() which makes a four-lab pool on which to run the MATLAB code in the file myscript.m. Use an appropriate filename, here named mylclbatch.m:
% FILENAME: mylclbatch.m
!echo "mylclbatch.m"
!hostname
pjob=batch('myscript','Profile','myslurmprofile','Pool',4,'CaptureDiary',true);
wait(pjob);
diary(pjob);
quit;
Prepare a job submission file with an appropriate filename, here named myjob.sub:
#!/bin/bash
# FILENAME: myjob.sub
echo "myjob.sub"
hostname
module load matlab
unset DISPLAY
matlab -nodisplay -r mylclbatch
Submit the job as a single compute node with one processor core.
One processor core runs myjob.sub and mylclbatch.m.
Once this job starts, a second job submission is made.
myjob.sub
< M A T L A B (R) >
Copyright 1984-2013 The MathWorks, Inc.
R2013a (8.1.0.604) 64-bit (glnxa64)
February 15, 2013
To get started, type one of these: helpwin, helpdesk, or demo.
For product information, visit www.mathworks.com.
mylclbatch.ma000.negishi.rcac.purdue.edu
SERIAL REGION: hostname:a000.negishi.rcac.purdue.edu
hostname numlabs labindex iteration
------------------------------- ------- -------- ---------
PARALLEL LOOP: a001.negishi.rcac.purdue.edu 4 1 2
PARALLEL LOOP: a002.negishi.rcac.purdue.edu 4 1 4
PARALLEL LOOP: a001.negishi.rcac.purdue.edu 4 1 5
PARALLEL LOOP: a002.negishi.rcac.purdue.edu 4 1 6
PARALLEL LOOP: a003.negishi.rcac.purdue.edu 4 1 1
PARALLEL LOOP: a003.negishi.rcac.purdue.edu 4 1 3
PARALLEL LOOP: a004.negishi.rcac.purdue.edu 4 1 7
PARALLEL LOOP: a004.negishi.rcac.purdue.edu 4 1 8
SERIAL REGION: hostname:a001.negishi.rcac.purdue.edu
Elapsed time in parallel loop: 5.411486
To scale up this method to handle a real application, increase the wall time in the submission command to accommodate a longer running job. Secondly, increase the wall time of myslurmprofile by using the Cluster Profile Manager in the Parallel menu to enter a new wall time in the property SubmitArguments.
For more information about MATLAB Parallel Computing Toolbox:
Parallel Toolbox (spmd)
The MATLAB Parallel Computing Toolbox (PCT) extends the MATLAB language with high-level, parallel-processing features such as parallel for loops, parallel regions, message passing, distributed arrays, and parallel numerical methods. It offers a shared-memory computing environment with a maximum of eight MATLAB workers (labs, threads; versions R2009a) and 12 workers (labs, threads; version R2011a) running on the local configuration in addition to your MATLAB client. Moreover, the MATLAB Distributed Computing Server (DCS) scales PCT applications up to the limit of your DCS licenses.
This section illustrates how to submit a small, parallel, MATLAB program with a parallel region (spmd statement) as a MATLAB pool job to a batch queue.
This example uses the submission command to submit to compute nodes a MATLAB client which interprets a Matlab .m with a user-defined cluster profile which scatters the MATLAB workers onto different compute nodes. This method uses the MATLAB interpreter, the Parallel Computing Toolbox, and the Distributed Computing Server; so, it requires and checks out six licenses: one MATLAB license for the client running on the compute node, one PCT license, and four DCS licenses. Four DCS licenses run the four copies of the spmd statement. This job is completely off the front end.
Prepare a MATLAB script called myscript.m:
% FILENAME: myscript.m
% SERIAL REGION
[c name] = system('hostname');
fprintf('SERIAL REGION: hostname:%s\n', name)
p = parpool('4');
fprintf(' hostname numlabs labindex\n')
fprintf(' ------------------------------- ------- --------\n')
tic;
% PARALLEL REGION
spmd
[c name] = system('hostname');
name = name(1:length(name)-1);
fprintf('PARALLEL REGION: %-31s %7d %8d\n', name,numlabs,labindex)
pause(2);
end
% SERIAL REGION
elapsed_time = toc; % get elapsed time in parallel region
delete(p);
fprintf('\n')
[c name] = system('hostname');
name = name(1:length(name)-1);
fprintf('SERIAL REGION: hostname:%s\n', name)
fprintf('Elapsed time in parallel region: %f\n', elapsed_time)
quit;
Prepare a job submission file with an appropriate filename, here named myjob.sub. Run with the name of the script:
#!/bin/bash
# FILENAME: myjob.sub
echo "myjob.sub"
module load matlab
unset DISPLAY
matlab -nodisplay -r myscript
Run MATLAB to set the default parallel configuration to your job configuration:
$ matlab -nodisplay
>> parallel.defaultClusterProfile('myslurmprofile');
>> quit;
$
Once this job starts, a second job submission is made.
myjob.sub
< M A T L A B (R) >
Copyright 1984-2011 The MathWorks, Inc.
R2011b (7.13.0.564) 64-bit (glnxa64)
August 13, 2011
To get started, type one of these: helpwin, helpdesk, or demo.
For product information, visit www.mathworks.com.
SERIAL REGION: hostname:a001.negishi.rcac.purdue.edu
Starting matlabpool using the 'myslurmprofile' profile ... connected to 4 labs.
hostname numlabs labindex
------------------------------- ------- --------
Lab 2:
PARALLEL REGION: a002.negishi.rcac.purdue.edu 4 2
Lab 1:
PARALLEL REGION: a001.negishi.rcac.purdue.edu 4 1
Lab 3:
PARALLEL REGION: a003.negishi.rcac.purdue.edu 4 3
Lab 4:
PARALLEL REGION: a004.negishi.rcac.purdue.edu 4 4
Sending a stop signal to all the labs ... stopped.
SERIAL REGION: hostname:a001.negishi.rcac.purdue.edu
Elapsed time in parallel region: 3.382151
Output shows the name of one compute node (a001) that processed the job submission file myjob.sub and the two serial regions. The job submission scattered four processor cores (four MATLAB labs) among four different compute nodes (a001,a002,a003,a004) that processed the four parallel regions. The total elapsed time demonstrates that the jobs ran in parallel.
For more information about MATLAB Parallel Computing Toolbox:
Distributed Computing Server (parallel job)
The MATLAB Parallel Computing Toolbox (PCT) enables a parallel job via the MATLAB Distributed Computing Server (DCS). The tasks of a parallel job are identical, run simultaneously on several MATLAB workers (labs), and communicate with each other. This section illustrates an MPI-like program.
This section illustrates how to submit a small, MATLAB parallel job with four workers running one MPI-like task to a batch queue. The MATLAB program broadcasts an integer to four workers and gathers the names of the compute nodes running the workers and the lab IDs of the workers.
This example uses the job submission command to submit a Matlab script with a user-defined cluster profile which scatters the MATLAB workers onto different compute nodes. This method uses the MATLAB interpreter, the Parallel Computing Toolbox, and the Distributed Computing Server; so, it requires and checks out six licenses: one MATLAB license for the client running on the compute node, one PCT license, and four DCS licenses. Four DCS licenses run the four copies of the parallel job. This job is completely off the front end.
Prepare a MATLAB script named myscript.m :
% FILENAME: myscript.m
% Specify pool size.
% Convert the parallel job to a pool job.
parpool('4');
spmd
if labindex == 1
% Lab (rank) #1 broadcasts an integer value to other labs (ranks).
N = labBroadcast(1,int64(1000));
else
% Each lab (rank) receives the broadcast value from lab (rank) #1.
N = labBroadcast(1);
end
% Form a string with host name, total number of labs, lab ID, and broadcast value.
[c name] =system('hostname');
name = name(1:length(name)-1);
fmt = num2str(floor(log10(numlabs))+1);
str = sprintf(['%s:%d:%' fmt 'd:%d '], name,numlabs,labindex,N);
% Apply global concatenate to all str's.
% Store the concatenation of str's in the first dimension (row) and on lab #1.
result = gcat(str,1,1);
if labindex == 1
disp(result)
end
end % spmd
matlabpool close force;
quit;
Also, prepare a job submission, here named myjob.sub. Run with the name of the script:
# FILENAME: myjob.sub
echo "myjob.sub"
module load matlab
unset DISPLAY
# -nodisplay: run MATLAB in text mode; X11 server not needed
# -r: read MATLAB program; use MATLAB JIT Accelerator
matlab -nodisplay -r myscript
Run MATLAB to set the default parallel configuration to your appropriate Profile:
$ matlab -nodisplay
>> defaultParallelConfig('myslurmprofile');
>> quit;
$
Submit the job as a single compute node with one processor core.
Once this job starts, a second job submission is made.
myjob.sub
< M A T L A B (R) >
Copyright 1984-2011 The MathWorks, Inc.
R2011b (7.13.0.564) 64-bit (glnxa64)
August 13, 2011
To get started, type one of these: helpwin, helpdesk, or demo.
For product information, visit www.mathworks.com.
>Starting matlabpool using the 'myslurmprofile' configuration ... connected to 4 labs.
Lab 1:
a006.negishi.rcac.purdue.edu:4:1:1000
a007.negishi.rcac.purdue.edu:4:2:1000
a008.negishi.rcac.purdue.edu:4:3:1000
a009.negishi.rcac.purdue.edu:4:4:1000
Sending a stop signal to all the labs ... stopped.
Did not find any pre-existing parallel jobs created by matlabpool.
Output shows the name of one compute node (a006) that processed the job submission file myjob.sub. The job submission scattered four processor cores (four MATLAB labs) among four different compute nodes (a006,a007,a008,a009) that processed the four parallel regions.
To scale up this method to handle a real application, increase the wall time in the submission command to accommodate a longer running job. Secondly, increase the wall time of myslurmprofile by using the Cluster Profile Manager in the Parallel menu to enter a new wall time in the property SubmitArguments.
For more information about parallel jobs:
Python
Notice: Python 2.7 has reached end-of-life on Jan 1, 2020 (announcement). Please update your codes and your job scripts to use Python 3.
Python is a high-level, general-purpose, interpreted, dynamic programming language. We suggest using Anaconda which is a Python distribution made for large-scale data processing, predictive analytics, and scientific computing. For example, to use the default Anaconda distribution:
$ module load conda
For a full list of available Anaconda and Python modules enter:
$ module spider conda
Example Python Jobs
This section illustrates how to submit a small Python job to a SLURM queue.
Link to section 'Example 1: Hello world' of 'Example Python Jobs' Example 1: Hello world
Prepare a Python input file with an appropriate filename, here named hello.py:
# FILENAME: hello.py
import string, sys
print("Hello, world!")
Prepare a job submission file with an appropriate filename, here named myjob.sub:
#!/bin/bash
# FILENAME: myjob.sub
module load conda
python hello.py
Hello, world!
Link to section 'Example 2: Matrix multiply' of 'Example Python Jobs' Example 2: Matrix multiply
Save the following script as matrix.py:
# Matrix multiplication program
x = [[3,1,4],[1,5,9],[2,6,5]]
y = [[3,5,8,9],[7,9,3,2],[3,8,4,6]]
result = [[sum(a*b for a,b in zip(x_row,y_col)) for y_col in zip(*y)] for x_row in x]
for r in result:
print(r)
Change the last line in the job submission file above to read:
python matrix.py
The standard output file from this job will result in the following matrix:
[28, 56, 43, 53]
[65, 122, 59, 73]
[63, 104, 54, 60]
Link to section 'Example 3: Sine wave plot using numpy and matplotlib packages' of 'Example Python Jobs' Example 3: Sine wave plot using numpy and matplotlib packages
Save the following script as sine.py:
import numpy as np
import matplotlib
matplotlib.use('Agg')
import matplotlib.pyplot as plt
x = np.linspace(-np.pi, np.pi, 201)
plt.plot(x, np.sin(x))
plt.xlabel('Angle [rad]')
plt.ylabel('sin(x)')
plt.axis('tight')
plt.savefig('sine.png')
Change your job submission file to submit this script and the job will output a png file and blank standard output and error files.
For more information about Python:
Managing Environments with Conda
Conda is a package manager in Anaconda that allows you to create and manage multiple environments where you can pick and choose which packages you want to use. To use Conda you must load an Anaconda module:
$ module load conda
Many packages are pre-installed in the global environment. To see these packages:
$ conda list
To create your own custom environment:
$ conda create --name MyEnvName python=3.8 FirstPackageName SecondPackageName -y
The --name option specifies that the environment created will be named MyEnvName. You can include as many packages as you require separated by a space. Including the -y option lets you skip the prompt to install the package. By default environments are created and stored in the $HOME/.conda directory.
To create an environment at a custom location:
$ conda create --prefix=$HOME/MyEnvName python=3.8 PackageName -y
To see a list of your environments:
$ conda env list
To remove unwanted environments:
$ conda remove --name MyEnvName --all
To add packages to your environment:
$ conda install --name MyEnvName PackageNames
To remove a package from an environment:
$ conda remove --name MyEnvName PackageName
Installing packages when creating your environment, instead of one at a time, will help you avoid dependency issues.
To activate or deactivate an environment you have created:
$ source activate MyEnvName
$ source deactivate MyEnvName
If you created your conda environment at a custom location using --prefix option, then you can activate or deactivate it using the full path.
$ source activate $HOME/MyEnvName
$ source deactivate $HOME/MyEnvName
To use a custom environment inside a job you must load the module and activate the environment inside your job submission script. Add the following lines to your submission script:
$ module load conda
$ source activate MyEnvName
For more information about Python:
Managing Packages with Pip
Pip is a Python package manager. Many Python package documentation provide pip instructions that result in permission errors because by default pip will install in a system-wide location and fail.
Exception:
Traceback (most recent call last):
... ... stack trace ... ...
OSError: [Errno 13] Permission denied: '/apps/cent7/anaconda/2020.07-py38/lib/python3.8/site-packages/mkl_random-1.1.1.dist-info'
If you encounter this error, it means that you cannot modify the global Python installation. We recommend installing Python packages in a conda environment. Detailed instructions for installing packages with pip can be found in our Python package installation page.
Below we list some other useful pip commands.
- Search for a package in PyPI channels:
$ pip search packageName - Check which packages are installed globally:
$ pip list - Check which packages you have personally installed:
$ pip list --user - Snapshot installed packages:
$ pip freeze > requirements.txt - You can install packages from a snapshot inside a new conda environment. Make sure to load the appropriate conda environment first.
$ pip install -r requirements.txt
For more information about Python:
Installing Packages
Installing Python packages in an Anaconda environment is recommended. One key advantage of Anaconda is that it allows users to install unrelated packages in separate self-contained environments. Individual packages can later be reinstalled or updated without impacting others. If you are unfamiliar with Conda environments, please check our Conda Guide.
To facilitate the process of creating and using Conda environments, we support a script (conda-env-mod) that generates a module file for an environment, as well as an optional Jupyter kernel to use this environment in a JupyterHub notebook.
You must load one of the anaconda modules in order to use this script.
$ module load conda
Step-by-step instructions for installing custom Python packages are presented below.
Link to section 'Step 1: Create a conda environment' of 'Installing Packages' Step 1: Create a conda environment
Users can use the conda-env-mod script to create an empty conda environment. This script needs either a name or a path for the desired environment. After the environment is created, it generates a module file for using it in future. Please note that conda-env-mod is different from the official conda-env script and supports a limited set of subcommands. Detailed instructions for using conda-env-mod can be found with the command conda-env-mod --help.
-
Example 1: Create a conda environment named mypackages in user's
$HOMEdirectory.$ conda-env-mod create -n mypackages -
Example 2: Create a conda environment named mypackages at a custom location.
$ conda-env-mod create -p /depot/mylab/apps/mypackagesPlease follow the on-screen instructions while the environment is being created. After finishing, the script will print the instructions to use this environment.
... ... ... Preparing transaction: ...working... done Verifying transaction: ...working... done Executing transaction: ...working... done +------------------------------------------------------+ | To use this environment, load the following modules: | | module load use.own | | module load conda-env/mypackages-py3.8.5 | +------------------------------------------------------+ Your environment "mypackages" was created successfully.
Note down the module names, as you will need to load these modules every time you want to use this environment. You may also want to add the module load lines in your jobscript, if it depends on custom Python packages.
By default, module files are generated in your $HOME/privatemodules directory. The location of module files can be customized by specifying the -m /path/to/modules option to conda-env-mod.
Note: The main differences between -p and -m are: 1) -p will change the location of packages to be installed for the env and the module file will still be located at the $HOME/privatemodules directory as defined in use.own. 2) -m will only change the location of the module file. So the method to load modules created with -m and -p are different, see Example 3 for details.
- Example 3: Create a conda environment named labpackages in your group's Data Depot space and place the module file at a shared location for the group to use.
$ conda-env-mod create -p /depot/mylab/apps/labpackages -m /depot/mylab/etc/modules ... ... ... Preparing transaction: ...working... done Verifying transaction: ...working... done Executing transaction: ...working... done +-------------------------------------------------------+ | To use this environment, load the following modules: | | module use /depot/mylab/etc/modules | | module load conda-env/labpackages-py3.8.5 | +-------------------------------------------------------+ Your environment "labpackages" was created successfully.
If you used a custom module file location, you need to run the module use command as printed by the command output above.
By default, only the environment and a module file are created (no Jupyter kernel). If you plan to use your environment in a JupyterHub notebook, you need to append a --jupyter flag to the above commands.
- Example 4: Create a Jupyter-enabled conda environment named labpackages in your group's Data Depot space and place the module file at a shared location for the group to use.
$ conda-env-mod create -p /depot/mylab/apps/labpackages -m /depot/mylab/etc/modules --jupyter ... ... ... Jupyter kernel created: "Python (My labpackages Kernel)" ... ... ... Your environment "labpackages" was created successfully.
Link to section 'Step 2: Load the conda environment' of 'Installing Packages' Step 2: Load the conda environment
-
The following instructions assume that you have used conda-env-mod script to create an environment named mypackages (Examples 1 or 2 above). If you used conda create instead, please use conda activate mypackages.
$ module load use.own $ module load conda-env/mypackages-py3.8.5Note that the conda-env module name includes the Python version that it supports (Python 3.8.5 in this example). This is same as the Python version in the conda module.
-
If you used a custom module file location (Example 3 above), please use module use to load the conda-env module.
$ module use /depot/mylab/etc/modules $ module load conda-env/labpackages-py3.8.5
Link to section 'Step 3: Install packages' of 'Installing Packages' Step 3: Install packages
Now you can install custom packages in the environment using either conda install or pip install.
Link to section 'Installing with conda' of 'Installing Packages' Installing with conda
-
Example 1: Install OpenCV (open-source computer vision library) using conda.
$ conda install opencv -
Example 2: Install a specific version of OpenCV using conda.
$ conda install opencv=4.5.5 -
Example 3: Install OpenCV from a specific anaconda channel.
$ conda install -c anaconda opencv
Link to section 'Installing with pip' of 'Installing Packages' Installing with pip
-
Example 4: Install pandas using pip.
$ pip install pandas -
Example 5: Install a specific version of pandas using pip.
$ pip install pandas==1.4.3Follow the on-screen instructions while the packages are being installed. If installation is successful, please proceed to the next section to test the packages.
Note: Do NOT run Pip with the --user argument, as that will install packages in a different location and might mess up your account environment.
Link to section 'Step 4: Test the installed packages' of 'Installing Packages' Step 4: Test the installed packages
To use the installed Python packages, you must load the module for your conda environment. If you have not loaded the conda-env module, please do so following the instructions at the end of Step 1.
$ module load use.own
$ module load conda-env/mypackages-py3.8.5
- Example 1: Test that OpenCV is available.
$ python -c "import cv2; print(cv2.__version__)" - Example 2: Test that pandas is available.
$ python -c "import pandas; print(pandas.__version__)"
If the commands finished without errors, then the installed packages can be used in your program.
Link to section 'Additional capabilities of conda-env-mod script' of 'Installing Packages' Additional capabilities of conda-env-mod script
The conda-env-mod tool is intended to facilitate creation of a minimal Anaconda environment, matching module file and optionally a Jupyter kernel. Once created, the environment can then be accessed via familiar module load command, tuned and expanded as necessary. Additionally, the script provides several auxiliary functions to help manage environments, module files and Jupyter kernels.
General usage for the tool adheres to the following pattern:
$ conda-env-mod help
$ conda-env-mod <subcommand> <required argument> [optional arguments]
where required arguments are one of
- -n|--name ENV_NAME (name of the environment)
- -p|--prefix ENV_PATH (location of the environment)
and optional arguments further modify behavior for specific actions (e.g. -m to specify alternative location for generated module files).
Given a required name or prefix for an environment, the conda-env-mod script supports the following subcommands:
- create - to create a new environment, its corresponding module file and optional Jupyter kernel.
- delete - to delete existing environment along with its module file and Jupyter kernel.
- module - to generate just the module file for a given existing environment.
- kernel - to generate just the Jupyter kernel for a given existing environment (note that the environment has to be created with a --jupyter option).
- help - to display script usage help.
Using these subcommands, you can iteratively fine-tune your environments, module files and Jupyter kernels, as well as delete and re-create them with ease. Below we cover several commonly occurring scenarios.
Note: When you try to use conda-env-mod delete, remember to include the arguments as you create the environment (i.e. -p package_location and/or -m module_location).
Link to section 'Generating module file for an existing environment' of 'Installing Packages' Generating module file for an existing environment
If you already have an existing configured Anaconda environment and want to generate a module file for it, follow appropriate examples from Step 1 above, but use the module subcommand instead of the create one. E.g.
$ conda-env-mod module -n mypackages
and follow printed instructions on how to load this module. With an optional --jupyter flag, a Jupyter kernel will also be generated.
Note that the module name mypackages should be exactly the same with the older conda environment name. Note also that if you intend to proceed with a Jupyter kernel generation (via the --jupyter flag or a kernel subcommand later), you will have to ensure that your environment has ipython and ipykernel packages installed into it. To avoid this and other related complications, we highly recommend making a fresh environment using a suitable conda-env-mod create .... --jupyter command instead.
Link to section 'Generating Jupyter kernel for an existing environment' of 'Installing Packages' Generating Jupyter kernel for an existing environment
If you already have an existing configured Anaconda environment and want to generate a Jupyter kernel file for it, you can use the kernel subcommand. E.g.
$ conda-env-mod kernel -n mypackages
This will add a "Python (My mypackages Kernel)" item to the dropdown list of available kernels upon your next login to the JupyterHub.
Note that generated Jupiter kernels are always personal (i.e. each user has to make their own, even for shared environments). Note also that you (or the creator of the shared environment) will have to ensure that your environment has ipython and ipykernel packages installed into it.
Link to section 'Managing and using shared Python environments' of 'Installing Packages' Managing and using shared Python environments
Here is a suggested workflow for a common group-shared Anaconda environment with Jupyter capabilities:
The PI or lab software manager:
-
Creates the environment and module file (once):
$ module purge $ module load conda $ conda-env-mod create -p /depot/mylab/apps/labpackages -m /depot/mylab/etc/modules --jupyter -
Installs required Python packages into the environment (as many times as needed):
$ module use /depot/mylab/etc/modules $ module load conda-env/labpackages-py3.8.5 $ conda install ....... # all the necessary packages
Lab members:
-
Lab members can start using the environment in their command line scripts or batch jobs simply by loading the corresponding module:
$ module use /depot/mylab/etc/modules $ module load conda-env/labpackages-py3.8.5 $ python my_data_processing_script.py ..... -
To use the environment in Jupyter notebooks, each lab member will need to create his/her own Jupyter kernel (once). This is because Jupyter kernels are private to individuals, even for shared environments.
$ module use /depot/mylab/etc/modules $ module load conda-env/labpackages-py3.8.5 $ conda-env-mod kernel -p /depot/mylab/apps/labpackages
A similar process can be devised for instructor-provided or individually-managed class software, etc.
Link to section 'Troubleshooting' of 'Installing Packages' Troubleshooting
- Python packages often fail to install or run due to dependency incompatibility with other packages. More specifically, if you previously installed packages in your home directory it is safer to clean those installations.
$ mv ~/.local ~/.local.bak $ mv ~/.cache ~/.cache.bak - Unload all the modules.
$ module purge - Clean up PYTHONPATH.
$ unset PYTHONPATH - Next load the modules (e.g. anaconda) that you need.
$ module load conda/2024.02-py311 $ module load use.own $ module load conda-env/2024.02-py311 - Now try running your code again.
- Few applications only run on specific versions of Python (e.g. Python 3.6). Please check the documentation of your application if that is the case.
Installing Packages from Source
We maintain several Anaconda installations. Anaconda maintains numerous popular scientific Python libraries in a single installation. If you need a Python library not included with normal Python we recommend first checking Anaconda. For a list of modules currently installed in the Anaconda Python distribution:
$ module load conda
$ conda list
# packages in environment at /apps/spack/bell/apps/anaconda/2020.02-py37-gcc-4.8.5-u747gsx:
#
# Name Version Build Channel
_ipyw_jlab_nb_ext_conf 0.1.0 py37_0
_libgcc_mutex 0.1 main
alabaster 0.7.12 py37_0
anaconda 2020.02 py37_0
...
If you see the library in the list, you can simply import it into your Python code after loading the Anaconda module.
If you do not find the package you need, you should be able to install the library in your own Anaconda customization. First try to install it with Conda or Pip. If the package is not available from either Conda or Pip, you may be able to install it from source.
Use the following instructions as a guideline for installing packages from source. Make sure you have a download link to the software (usually it will be a tar.gz archive file). You will substitute it on the wget line below.
We also assume that you have already created an empty conda environment as described in our Python package installation guide.
$ mkdir ~/src
$ cd ~/src
$ wget http://path/to/source/tarball/app-1.0.tar.gz
$ tar xzvf app-1.0.tar.gz
$ cd app-1.0
$ module load conda
$ module load use.own
$ module load conda-env/mypackages-py3.8.5
$ python setup.py install
$ cd ~
$ python
>>> import app
>>> quit()
The "import app" line should return without any output if installed successfully. You can then import the package in your python scripts.
If you need further help or run into any issues installing a library, contact us or drop by Coffee Hour for in-person help.
For more information about Python:
Example: Create and Use Biopython Environment with Conda
Link to section 'Using conda to create an environment that uses the biopython package' of 'Example: Create and Use Biopython Environment with Conda' Using conda to create an environment that uses the biopython package
To use Conda you must first load the anaconda module:
module load conda
Create an empty conda environment to install biopython:
conda-env-mod create -n biopython
Now activate the biopython environment:
module load use.own
module load conda-env/biopython-py3.12.5
Install the biopython packages in your environment:
conda install --channel anaconda biopython -y
Fetching package metadata ..........
Solving package specifications .........
.......
Linking packages ...
[ COMPLETE ]|################################################################
The --channel option specifies that it searches the anaconda channel for the biopython package. The -y argument is optional and allows you to skip the installation prompt. A list of packages will be displayed as they are installed.
Remember to add the following lines to your job submission script to use the custom environment in your jobs:
module load conda
module load use.own
module load conda-env/biopython-py3.12.5
If you need further help or run into any issues with creating environments, contact us or drop by Coffee Hour for in-person help.
For more information about Python:
Numpy Parallel Behavior
The widely available Numpy package is the best way to handle numerical computation in Python. The numpy package provided by our anaconda modules is optimized using Intel's MKL library. It will automatically parallelize many operations to make use of all the cores available on a machine.
In many contexts that would be the ideal behavior. On the cluster however that very likely is not in fact the preferred behavior because often more than one user is present on the system and/or more than one job on a node. Having multiple processes contend for those resources will actually result in lesser performance.
Setting the MKL_NUM_THREADS or OMP_NUM_THREADS environment variable(s) allows you to control this behavior. Our anaconda modules automatically set these variables to 1 if and only if you do not currently have that variable defined.
When submitting batch jobs it is always a good idea to be explicit rather than implicit. If you are submitting a job that you want to make use of the full resources available on the node, set one or both of these variables to the number of cores you want to allow numpy to make use of.
#!/bin/bash
module load conda
export MKL_NUM_THREADS=128
...
If you are submitting multiple jobs that you intend to be scheduled together on the same node, it is probably best to restrict numpy to a single core.
#!/bin/bash
module load conda
export MKL_NUM_THREADS=1
R
R, a GNU project, is a language and environment for data manipulation, statistics, and graphics. It is an open source version of the S programming language. R is quickly becoming the language of choice for data science due to the ease with which it can produce high quality plots and data visualizations. It is a versatile platform with a large, growing community and collection of packages.
For more general information on R visit The R Project for Statistical Computing.
Running R jobs
This section illustrates how to submit a small R job to a SLURM queue. The example job computes a Pythagorean triple.
Prepare an R input file with an appropriate filename, here named myjob.R:
# FILENAME: myjob.R
# Compute a Pythagorean triple.
a = 3
b = 4
c = sqrt(a*a + b*b)
c # display result
Prepare a job submission file with an appropriate filename, here named myjob.sub:
#!/bin/bash
# FILENAME: myjob.sub
module load r
# --vanilla:
# --no-save: do not save datasets at the end of an R session
R --vanilla --no-save < myjob.R
For other examples or R jobs:
Installing R packages
Link to section 'Challenges of Managing R Packages in the Cluster Environment' of 'Installing R packages' Challenges of Managing R Packages in the Cluster Environment
- Different clusters have different hardware and softwares. So, if you have access to multiple clusters, you must install your R packages separately for each cluster.
- Each cluster has multiple versions of R and packages installed with one version of R may not work with another version of R. So, libraries for each R version must be installed in a separate directory.
- You can define the directory where your R packages will be installed using the environment variable
R_LIBS_USER. - For your convenience, a sample ~/.Rprofile example file is provided that can be downloaded to your cluster account and renamed into
~/.Rprofile(or appended to one) to customize your installation preferences. Detailed instructions.
Link to section 'Installing Packages' of 'Installing R packages' Installing Packages
-
Step 0: Set up installation preferences.
Follow the steps for setting up your~/.Rprofilepreferences. This step needs to be done only once. If you have created a~/.Rprofilefile previously on Negishi, ignore this step. -
Step 1: Check if the package is already installed.
As part of the R installations on community clusters, a lot of R libraries are pre-installed. You can check if your package is already installed by opening an R terminal and entering the commandinstalled.packages(). For example,module load r/4.4.1 Rinstalled.packages()["units",c("Package","Version")] Package Version "units" "0.8-1" quit()If the package you are trying to use is already installed, simply load the library, e.g.,
library('units'). Otherwise, move to the next step to install the package. -
Step 2: Load required dependencies. (if needed)
For simple packages you may not need this step. However, some R packages depend on other libraries. For example, thesfpackage depends ongdalandgeoslibraries. So, you will need to load the corresponding modules before installingsf. Read the documentation for the package to identify which modules should be loaded.module load gdal module load geos -
Step 3: Install the package.
Now install the desired package using the commandinstall.packages('package_name'). R will automatically download the package and all its dependencies from CRAN and install each one. Your terminal will show the build progress and eventually show whether the package was installed successfully or not.Rinstall.packages('sf', repos="https://cran.case.edu/") Installing package into ‘/home/myusername/R/x86_64-pc-linux-gnu-library/4.4.1’ (as ‘lib’ is unspecified) trying URL 'https://cran.case.edu/src/contrib/sf_0.9-7.tar.gz' Content type 'application/x-gzip' length 4203095 bytes (4.0 MB) ================================================== downloaded 4.0 MB ... ... more progress messages ... ... ** testing if installed package can be loaded from final location ** testing if installed package keeps a record of temporary installation path * DONE (sf) The downloaded source packages are in ‘/tmp/RtmpSVAGio/downloaded_packages’ - Step 4: Troubleshooting. (if needed)
If Step 3 ended with an error, you need to investigate why the build failed. Most common reason for build failure is not loading the necessary modules.
Link to section 'Loading Libraries' of 'Installing R packages' Loading Libraries
Once you have packages installed you can load them with the library() function as shown below:
library('packagename')
The package is now installed and loaded and ready to be used in R.
Link to section 'Example: Installing dplyr' of 'Installing R packages' Example: Installing dplyr
The following demonstrates installing the dplyr package assuming the above-mentioned custom ~/.Rprofile is in place (note its effect in the "Installing package into" information message):
module load r
R
install.packages('dplyr', repos="http://ftp.ussg.iu.edu/CRAN/")
Installing package into ‘/home/myusername/R/negishi/4.4.1’
(as ‘lib’ is unspecified)
...
also installing the dependencies 'crayon', 'utf8', 'bindr', 'cli', 'pillar', 'assertthat', 'bindrcpp', 'glue', 'pkgconfig', 'rlang', 'Rcpp', 'tibble', 'BH', 'plogr'
...
...
...
The downloaded source packages are in
'/tmp/RtmpHMzm9z/downloaded_packages'
library(dplyr)
Attaching package: 'dplyr'
For more information about installing R packages:
Loading Data into R
R is an environment for manipulating data. In order to manipulate data, it must be brought into the R environment. R has a function to read any file that data is stored in. Some of the most common file types like comma-separated variable(CSV) files have functions that come in the basic R packages. Other less common file types require additional packages to be installed. To read data from a CSV file into the R environment, enter the following command in the R prompt:
> read.csv(file = "path/to/data.csv", header = TRUE)
When R reads the file it creates an object that can then become the target of other functions. By default the read.csv() function will give the object the name of the .csv file. To assign a different name to the object created by read.csv enter the following in the R prompt:
> my_variable <- read.csv(file = "path/to/data.csv", header = FALSE)
To display the properties (structure) of loaded data, enter the following:
> str(my_variable)
For more functions and tutorials:
RStudio
RStudio is a graphical integrated development environment (IDE) for R. RStudio is the most popular environment for developing both R scripts and packages. RStudio is provided on most Research systems.
There are two methods to launch RStudio on the cluster: command-line and application menu icon.
Link to section 'Launch RStudio by the command-line:' of 'RStudio' Launch RStudio by the command-line:
module load gcc
module load r
module load rstudio
rstudio
Note that RStudio is a graphical program and in order to run it you must have a local X11 server running or use Thinlinc Remote Desktop environment. See the ssh X11 forwarding section for more details.
Link to section 'Launch Rstudio by the application menu icon:' of 'RStudio' Launch Rstudio by the application menu icon:
- Log into desktop.negishi.rcac.purdue.edu with web browser or ThinLinc client
- Click on the
Applicationsdrop down menu on the top left corner - Choose
Cluster Softwareand thenRStudio
![]()
R and RStudio are free to download and run on your local machine. For more information about RStudio:
Setting Up R Preferences with .Rprofile
For your convenience, a sample ~/.Rprofile example file is provided that can be downloaded to your cluster account and renamed into ~/.Rprofile (or appended to one). Follow these steps to download our recommended ~/.Rprofile example and copy it into place:
curl -#LO https://www.rcac.purdue.edu/files/knowledge/run/examples/apps/r/Rprofile_example
mv -ib Rprofile_example ~/.Rprofile
The above installation step needs to be done only once on Negishi. Now load the R module and run R:
module load r/4.4.1
R
.libPaths()
[1] "/home/myusername/R/negishi/4.1.2-gcc-6.3.0-ymdumss"
[2] "/apps/spack/negishi/apps/r/4.1.2-gcc-6.3.0-ymdumss/rlib/R/library"
.libPaths() should output something similar to above if it is set up correctly.
You are now ready to install R packages into the dedicated directory /home/myusername/R/negishi/4.1.2-gcc-6.3.0-ymdumss.
Singularity
On Negishi, Singularity functionality is provided by Apptainer - see Apptainer section for details.
Windows
Windows virtual machines (VMs) are supported as batch jobs on HPC systems. This section illustrates how to submit a job and run a Windows instance in order to run Windows applications on the high-performance computing systems.
The following images are pre-configured and made available by staff:
- Windows 2016 Server Basic (minimal software pre-loaded)
- Windows 2016 Server GIS (GIS Software Stack pre-loaded)
The Windows VMs can be launched in two fashions:
- Menu Launcher - Point and click to start
- Command Line - Advanced and customized usage
Click each of the above links for detailed instructions on using them.
Link to section 'Software Provided in Pre-configured Virtual Machines' of 'Windows' Software Provided in Pre-configured Virtual Machines
The Windows 2016 Base server image available on Negishi has the following software packages preloaded:
- Anaconda Python 2 and Python 3
- JMP 13
- Matlab R2017b
- Microsoft Office 2016
- Notepad++
- NVivo 12
- Rstudio
- Stata SE 15
- VLC Media Player
Command line
If you wish to work with Windows VMs on the command line or work into scripted workflows you can interact directly with the Windows system:
Copy a Windows 2016 Server VM image to your storage. Scratch or Research Data Depot are good locations to save a VM image. If you are using scratch, remember that scratch spaces are temporary, and be sure to safely back up your disk image somewhere permanent, such as Research Data Depot or Fortress. To copy a basic image:
$ cp /apps/external/apps/windows/images/latest.qcow2 $RCAC_SCRATCH/windows.qcow2
To copy a GIS image:
$ cp /depot/itap/windows/gis/2k16.qcow2 $RCAC_SCRATCH/windows.qcow2
To launch a virtual machine in a batch job, use the "windows" script, specifying the path to your Windows virtual machine image. With no other command-line arguments, the windows script will autodetect a number cores and memory for the Windows VM. A Windows network connection will be made to your home directory. To launch:
$ windows -i $RCAC_SCRATCH/windows.qcow2
Link to section 'Command line options:' of 'Command line' Command line options:
-i <path to qcow image file> (For example, $RCAC_SCRATCH/windows-2k16.qcow2)
-m <RAM>G (For example, 32G)
-c <cores> (For example, 20)
-s <smbpath> (UNIX Path to map as a drive, for example, $RCAC_SCRATCH)
-b (If present, launches VM in background. Use VNC to connect to Windows.)
To launch a virtual machine with 32GB of RAM, 20 cores, and a network mapping to your home directory:
$ windows -i /path/to/image.qcow2 -m 32G -c 20 -s $HOME
To launch a virtual machine with 16GB of RAM, 10 cores, and a network mapping to your Data Depot space:
$ windows -i /path/to/image.qcow2 -m 16G -c 10 -s /depot/mylab
The Windows 2016 server desktop will open, and automatically log in as an administrator, so that you can install any software into the Windows virtual machine that your research requires. Changes to the image will be stored in the file specified with the -i option.
Menu Launcher
Windows VMs can be easily launched through the login/thinlinc">Thinlinc remote desktop environment.
- Log in via login/thinlinc">Thinlinc.
- Click on Applications menu in the upper left corner.
- Look under the Cluster Software menu.
- The "Windows 10" launcher will launch a VM directly on the front-end.
- Follow the dialogs to set up your VM.
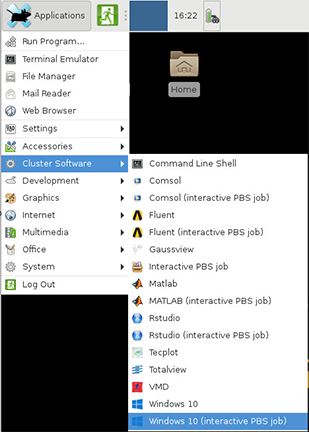
The dialog menus will walk you through setting up and loading your VM.
- You can choose to create a new image or load a saved image.
- New VMs should be saved on Scratch or Research Data Depot as they are too large for Home Directories.
- If you are using scratch, remember that scratch spaces are temporary, and be sure to safely back up your disk image somewhere permanent, such as Research Data Depot or Fortress.
You will also be prompted to select a storage space to mount on your image (Home, Scratch, or Data Depot). You can only choose one to be mounted. It will appear on a shortcut on the desktop once the VM loads.
Link to section 'Notes' of 'Menu Launcher' Notes
Using the menu launcher will launch automatically select reasonable CPU and memory values. If you wish to choose other options or work Windows VMs into scripted workflows see the section on using the command line.
BioContainers Collection
Link to section 'What is BioContainers?' of 'BioContainers Collection' What is BioContainers?
The BioContainers project came from the idea of using the containers-based technologies such as Docker or rkt for bioinformatics software. Having a common and controllable environment for running software could help to deal with some of the current problems during software development and distribution. BioContainers is a community-driven project that provides the infrastructure and basic guidelines to create, manage and distribute bioinformatics containers with a special focus on omics fields such as proteomics, genomics, transcriptomics and metabolomics. . For more information, please visit BioContainers project.
Link to section ' Getting Started ' of 'BioContainers Collection' Getting Started
Users can download bioinformatic containers from the BioContainers.pro and run them directly using Singularity instructions from the corresponding container’s catalog page.
Brief Singularity guide and examples are available at the Negishi Singularity user guide page. Detailed Singularity user guide is available at: sylabs.io/guides/3.8/user-guide
In addition, a subset of pre-downloaded biocontainers wrapped into convenient software modules are provided. These modules wrap underlying complexity and provide the same commands that are expected from non-containerized versions of each application.
On Negishi, type the command below to see the lists of biocontainers we deployed.
module load biocontainers
module avail
------------ BioContainers collection modules -------------
bamtools/2.5.1
beast2/2.6.3
bedtools/2.30.0
blast/2.11.0
bowtie2/2.4.2
bwa/0.7.17
cufflinks/2.2.1
deeptools/3.5.1
fastqc/0.11.9
faststructure/1.0
htseq/0.13.5
[....]Link to section ' Example ' of 'BioContainers Collection' Example
This example demonstrates how to run BLASTP with the blast module. This blast module is a biocontainer wrapper for NCBI BLAST.
module load biocontainers
module load blast
blastp -query query.fasta -db nr -out output.txt -outfmt 6 -evalue 0.01 To run a job in batch mode, first prepare a job script that specifies the BioContainer modules you want to launch and the resources required to run it. Then, use the sbatch command to submit your job script to Slurm. The following example shows the job script to use Bowtie2 in bioinformatic analysis.
#!/bin/bash
#SBATCH -A myqueuename
#SBATCH -o bowtie2_%j.txt
#SBATCH -e bowtie2_%j.err
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=8
#SBATCH --time=1:30:00
#SBATCH --job-name bowtie2
# Load the Bowtie module
module load biocontainers
module load bowtie2
# Indexing a reference genome
bowtie2-build ref.fasta ref
# Aligning paired-end reads
bowtie2 -p 8 -x ref -1 reads_1.fq -2 reads_2.fq -S align.sam To help users get started, we provided detailed user guides for each containerized bioinformatics module on the ReadTheDocs platform
Ansys Fluent
Ansys is a CAE/multiphysics engineering simulation software that utilizes finite element analysis for numerically solving a wide variety of mechanical problems. The software contains a list of packages and can simulate many structural properties such as strength, toughness, elasticity, thermal expansion, fluid dynamics as well as acoustic and electromagnetic attributes.
Link to section 'Ansys Licensing' of 'Ansys Fluent' Ansys Licensing
The Ansys licensing on our community clusters is maintained by Purdue ECN group. There are two types of licenses: teaching and research. For more information, please refer to ECN Ansys licensing page. If you are interested in purchasing your own research license, please send email to software@ecn.purdue.edu.
Link to section 'Ansys Workflow' of 'Ansys Fluent' Ansys Workflow
Ansys software consists of several sub-packages such as Workbench and Fluent. Most simulations are performed using the Ansys Workbench console, a GUI interface to manage and edit the simulation workflow. It requires X11 forwarding for remote display so a SSH client software with X11 support or a remote desktop portal is required. Please see Logging In section for more details. To ensure preferred performance, ThinLinc remote desktop connection is highly recommended.
Typically users break down larger structures into small components in geometry with each of them modeled and tested individually. A user may start by defining the dimensions of an object, adding weight, pressure, temperature, and other physical properties.
Ansys Fluent is a computational fluid dynamics (CFD) simulation software known for its advanced physics modeling capabilities and accuracy. Fluent offers unparalleled analysis capabilities and provides all the tools needed to design and optimize new equipment and to troubleshoot existing installations.
In the following sections, we provide step-by-step instructions to lead you through the process of using Fluent. We will create a classical elbow pipe model and simulate the fluid dynamics when water flows through the pipe. The project files have been generated and can be downloaded via fluent_tutorial.zip.
Link to section 'Loading Ansys Module' of 'Ansys Fluent' Loading Ansys Module
Different versions of Ansys are installed on the clusters and can be listed with module spider or module avail command in the terminal.
$ module avail ansys/
---------------------- Core Applications -----------------------------
ansys/2019R3 ansys/2020R1 ansys/2021R2 ansys/2022R1 (D)Before launching Ansys Workbench, a specific version of Ansys module needs to be loaded. For example, you can module load ansys/2021R2 to use the latest Ansys 2021R2. If no version is specified, the default module -> (D) (ansys/2022R1 in this case) will be loaded. You can also check the loaded modules with module list command.
Link to section 'Launching Ansys Workbench' of 'Ansys Fluent' Launching Ansys Workbench
Open a terminal on Negishi, enter rcac-runwb2 to launch Ansys Workbench.
You can also use runwb2 to launch Ansys Workbench. The main difference between runwb2and rcac-runwb2 is that the latter sets the project folder to be in your scratch space. Ansys has an known bug that it might crash when the project folder is set to $HOME on our systems.
Preparing Case Files for Fluent
Link to section 'Creating a Fluent fluid analysis system' of 'Preparing Case Files for Fluent' Creating a Fluent fluid analysis system
In the Ansys Workbench, create a new fluid flow analysis by double-clicking the Fluid Flow (Fluent) option under the Analysis Systems in the Toolbox on the left panel. You can also drag-and-drop the analysis system into the Project Schematic. A green dotted outline indicating a potential location for the new system initially appears in the Project Schematic. When you drag the system to one of the outlines, it turns into a red box to indicate the chosen location of the new system.
The red rectangle indicates the Fluid Flow system for Fluent, which includes all the essential workflows from “2 Geometry” to “6 Results”. You can rename it and carry out the necessary step-by-step procedures by double-clicking the corresponding cells.
It is important to save the project. Ansys Workbench saves the project with a .wbpj extension and also all the supporting files into a folder with the same name. In this case, a file named elbow_demo.wbpj and a folder $Ansys_PROJECT_FOLDER/elbow_demo_files/ are created in the Ansys project folder:
$ ll
total 33
drwxr-xr-x 7 myusername itap 9 Mar 3 17:47 elbow_demo_files
-rw-r--r-- 1 myusername itap 42597 Mar 3 17:47 elbow_demo.wbpj
You should always “Update Project” and save it after finishing a procedure.
Link to section 'Creating Geometry in the Ansys DesignModeler' of 'Preparing Case Files for Fluent' Creating Geometry in the Ansys DesignModeler
Create a geometry in the Ansys DesignModeler (by double-clicking “Geometry” cell in workflow), or import the appropriate geometry file (by right-clicking the Geometry cell and selecting “Import Geometry” option from the context menu).
You can use Ansys DesignModeler to create 2D/3D geometries or even draw the objects yourself. In our example, we created only half of the elbow pipe because the symmetry of the structure is taken into account to reduce the computation intensity.
After saving the geometry, a geometry file FFF.agdb will be created in the folder: $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/FFF/DM/. The project in Workbench will be updated automatically.
If you import a pre-existing geometry into Ansys DesignModeler, it will also generate this file with the same filename at this location.
Link to section 'Creating mesh in the Ansys Meshing' of 'Preparing Case Files for Fluent' Creating mesh in the Ansys Meshing
Now that we have created the elbow pipe geometry, a computational mesh can be generated by the Meshing application throughout the flow volume.
With the successful creation of the geometry, there should be a green check showing the completion of “Geometry” in the Ansys Workbench. A Refresh Required icon within the “Mesh” cell indicates the mesh needs to be updated and refreshed for the system.
Then it’s time to open the Ansys Meshing application by double-clicking the “Mesh” cell and editing the mesh for the project. Generally, there are several steps we need to take to define the mesh:
- Create names for all geometry boundaries such as the inlets, outlets and fluid body. Note: You can use the strings “velocity inlet” and “pressure outlet” in the named selections (with or without hyphens or underscore characters) to allow Ansys Fluent to automatically detect and assign the corresponding boundary types accordingly. Use “Fluid” for the body to let Ansys Fluent automatically detect that the volume is a fluid zone and treat it accordingly.
- Set basic meshing parameters for the Ansys Meshing application. Here are several important parameters you may need to assign: Sizing, Quality, Body Sizing Control, Inflation.
- Select “Generate” to generate the mesh and “Update” to update the mesh into the system. Note: Once the mesh is generated, you can view the mesh statistics by opening the Statistics node in the Details of “Mesh” view. This will display information such as the number of nodes and the number of elements, which gives you a general idea for the future computational resources and time.
After generation and updating the mesh, a mesh file FFF.msh will be generated in folder $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/FFF/MECH/ and a mesh database file FFF.mshdb will be generated in folder $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/global/MECH/.
Parameters used in demo case (use default if not assigned):
- Length Unit=”mm”
- Names defined for geometry:
- velocity-inlet-large (large inlet on pipe);
- velocity-inlet-small (small inlet on pipe);
- pressure-outlet (outlet on pipe);
- symmetry (symmetry surface);
- Fluid (body);
- Mesh:
- Quality: Smoothing=”high”;
- Inflation: Use Automatic Inflation=“Program Controlled”, Inflation Option=”Smooth Transition”;
- Statistics:
- Nodes=29371;
- Elements=87647.
Link to section 'Calculation with Fluent' of 'Preparing Case Files for Fluent' Calculation with Fluent
Now all the preparations have been ready for the numerical calculation in Ansys Fluent. Both “Geometry” and “Mesh” cells should have green checks on. We can set up the CFD simulation parameters in Ansys Fluent by double-clicking the “Setup” cell.
When Ansys Fluent is first started or by selecting “editing” on the “Setup” cell, the Fluent Launcher is displayed, enabling you to view and/or set certain Ansys Fluent start-up options (e.g. Precision, Parallel, Display). Note that “Dimension” is fixed to “3D” because we are using a 3D model in this project.
After the Fluent is opened, an Ansys Fluent settings file FFF.set is written under the folder $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/FFF/Fluent/.
Then we are going to set up all the necessary parameters for Fluent computation. Here are the key steps for the setup:
- Setting up the domain:
- Change the units for length to be consistent with the Mesh;
- Check the mesh statistics and quality;
- Setting up physics:
- Solver: “Energy”, “Viscous Model”, “Near-Wall Treatment”;
- Materials;
- Zones;
- Boundaries: Inlet, Outlet, Internal, Symmetry, Wall;
- Solving:
- Solution Methods;
- Reports;
- Initialization;
- Iterations and output frequency.
Then the calculation will be carried out and the results will be written out into FFF-1.cas.gz under folder $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/FFF/Fluent/.
This file contains all the settings and simulation results which can be loaded for post analysis and re-computation (more details will be introduced in the following sections). If only configurations and settings within the Fluent are needed, we can open independent Fluent or submit Fluent jobs with bash commands by loading the existing case in order to facilitate the computation process.
Parameters used in demo case (use default if not assigned):
- Domain Setup: Length Units=”mm”;
- Solver: Energy=”on”; Viscous Model=”k-epsilon”; Near-Wall Treatment=”Enhanced Wall Treatment”;
- Materials: water (Density=1000[kg/m^3]; Specific Heat=4216[J/kg-k]; Thermal Conductivity=0.677[w/m-k]; Viscosity=8e-4[kg/m-s]);
- Zones=”fluid (water)”;
- Inlet=”velocity-inlet-large” (Velocity Magnitude=0.4m/s, Specification Method=”Intensity and Hydraulic Diameter”, Turbulent Intensity=5%; Hydraulic Diameter=100mm; Thermal Temperature=293.15k) &”velocity-inlet-small” (Velocity Magnitude=1.2m/s, Specification Method=”Intensity and Hydraulic Diameter”, Turbulent Intensity=5%; Hydraulic Diameter=25mm; Thermal Temperature=313.15k); Internal=”interior-fluid”; Symmetry=”symmetry”; Wall=”wall-fluid”;
- Solution Methods: Gradient=”Green-Gauss Node Based”;
- Report: plot residual and “Facet Maximum” for “pressure-outlet”
- Hybrid Initialization;
- 300 iterations.
Case Calculating with Fluent
Link to section 'Calculation with Fluent' of 'Case Calculating with Fluent' Calculation with Fluent
Now all the files are ready for the Fluent calculations. Both “Geometry” and “Mesh” cells should have green checks. We can set up the CFD simulation parameters in the Ansys Fluent by double-clicking the “Setup” cell.
Ansys Fluent Launcher can be started by selecting “editing” on the “Setup” cell with many startup options (e.g. Precision, Parallel, Display). Note that “Dimension” is fixed to “3D” because we are using a 3D model in this project.
After the Fluent is opened, an Ansys Fluent settings file FFF.set is written under the folder $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/FFF/Fluent/.
Then we are going to set up all the necessary parameters for Fluent computation. Here are the key steps for the setup:
- Setting up the domain:
- Change the units for length to be consistent with the Mesh;
- Check the mesh statistics and quality;
- Setting up physics:
- Solver: “Energy”, “Viscous Model”, “Near-Wall Treatment”;
- Materials;
- Zones;
- Boundaries: Inlet, Outlet, Internal, Symmetry, Wall;
- Solving:
- Solution Methods;
- Reports;
- Initialization;
- Iterations and output frequency.
Then the calculation will be carried out and the results will be written out into FFF-1.cas.gz under folder $Ansys_PROJECT_FOLDER/elbow_demo_file/dp0/FFF/Fluent/.
This file contains all the settings and simulation results which can be loaded for post analysis and re-computation (more details will be introduced in the following sections). If only configurations and settings within the Fluent are needed, we can open independent Fluent or submit Fluent jobs with bash commands by loading the existing case in order to facilitate the computation process.
Parameters used in demo case (use default if not assigned):
- Domain Setup: Length Units=”mm”;
- Solver: Energy=”on”; Viscous Model=”k-epsilon”; Near-Wall Treatment=”Enhanced Wall Treatment”;
- Materials: water (Density=1000[kg/m^3]; Specific Heat=4216[J/kg-k]; Thermal Conductivity=0.677[w/m-k]; Viscosity=8e-4[kg/m-s]);
- Zones=”fluid (water)”;
- Inlet=”velocity-inlet-large” (Velocity Magnitude=0.4m/s, Specification Method=”Intensity and Hydraulic Diameter”, Turbulent Intensity=5%; Hydraulic Diameter=100mm; Thermal Temperature=293.15k) &”velocity-inlet-small” (Velocity Magnitude=1.2m/s, Specification Method=”Intensity and Hydraulic Diameter”, Turbulent Intensity=5%; Hydraulic Diameter=25mm; Thermal Temperature=313.15k); Internal=”interior-fluid”; Symmetry=”symmetry”; Wall=”wall-fluid”;
- Solution Methods: Gradient=”Green-Gauss Node Based”;
- Report: plot residual and “Facet Maximum” for “pressure-outlet”
- Hybrid Initialization;
- 300 iterations.
Link to section 'Results analysis' of 'Case Calculating with Fluent' Results analysis
The best methods to view and analyze the simulation should be the Ansys Fluent (directly after computation) or the Ansys CFD-Post (entering “Results” in Ansys Workbench). Both methods are straightforward so we will not cover this part in this tutorial. Here is a final simulation result showing the temperature of the symmetry after 300 iterations for reference:

Fluent Text User Interface and Journal File
Link to section 'Fluent Text User Interface (TUI)' of 'Fluent Text User Interface and Journal File' Fluent Text User Interface (TUI)
If you pay attention to the “Console” window in the Fluent window when setting up and carrying out the calculation, corresponding commands can be found and executed one after another. Almost all the setting processes can be accomplished by the command lines, which is called Fluent Text User Interface (TUI). Here are the main commands in Fluent TUI:
adjoint/ parallel/ solve/
define/ plot/ surface/
display/ preferences/ turbo-workflow/
exit print-license-usage views/
file/ report/
mesh/ server/
For example, instead of opening a case by clicking buttons in Ansys Fluent, we can type /file read-case case_file_name.cas.gz to open the saved case.
Link to section 'Fluent Journal Files' of 'Fluent Text User Interface and Journal File' Fluent Journal Files
A Fluent journal file is a series of TUI commands stored in a text file. The file can be written in a text editor or generated by Fluent as a transcript of the commands given to Fluent during your session.
A journal file generated by Fluent will include any GUI operations (in a TUI form, though). This is quite useful if you have a series of tasks that you need to execute, as it provides a shortcut. To record a journal file, start recording with File -> Write -> Start Journal..., perform whatever tasks you need, and then stop recording with File -> Write -> Stop Journal...
You can also write your own journal file into a text file. The basic rule for a Fluent journal file is to reproduce the TUI commands that controlled the configuration and calculation of Fluent in their order. You can add a comment in a line starting with a ; (semicolon).
Here are some reasons why you should use a Fluent journal file:
- Using journal files with bash scripting can allow you to automate your jobs.
- Using journal files can allow you to parameterize your models easily and automatically.
- Using a journal file can set parameters you do not have in your case file e.g. autosaving.
- Using a journal file can allow you to safely save, stop and restart your jobs easily.
The order of your journal file commands is highly important. The correct sequences must be followed and some stages have multiple options e.g. different initialization methods.
Here is a sample Fluent journal file for the demo case:
;testJournal.jou
;Set the TUI version for Fluent
/file/set-tui-version "22.1"
;Read the case. The default folder
/file read-case /home/jin456/Fluent_files/tutorial_case1/elbow_files/dp0/FFF/Fluent/FFF-1.cas.gz
;Initialize the case with Hybrid Initialization
/solve/initialize/hyb-initialization
;Set Number of Iterations to 1000, Reporting Interval to 10 iterations and Profile Update Interval to 1 iteration
/solve/iterate 1000 10 1
;Outputting solver performance data upon completion of the simulation
/parallel timer usage
;Write out the simulation results.
/file write-case-data /home/jin456/Fluent_files/tutorial_case1/elbow_files/dp0/FFF/Fluent/result.cas.h5
;After computation, exit Flent
/exit
Before running this Fluent journal file, you need to make sure: 1) the ansys module has been loaded (it’s highly recommended to load the same version of Ansys when you built the case project); 2) the project case file (***.cas.gz) has been created.
Then we can use Fluent to run this journal file by simply using:fluent 3ddp -t$NTASKS -g -i testJournal.jou in the terminal. Here, 3d indicates this is a 3d model, dp indicates double precision, -t$NTASKS tells Fluent how many Solver Processes it will take (e.g. -t4), -g means to run without the GUI or graphics, -i testJournal.jou tells Fluent to read the specific journal file.
Here is a table for the available command line Options for Linux/UNIX and Windows Platforms in Ansys Fluent.
| Option | Platform | Description |
|---|---|---|
-cc |
all | Use the classic color scheme |
-ccp x |
Windows only | Use the Microsoft Job Scheduler where x is the head node name. |
-cnf=x |
all | Specify the hosts or machine list file |
-driver |
all | Sets the graphics driver (available drivers vary by platform - opengl or x11 or null(Linux/UNIX) - opengl or msw or null (Windows)) |
-env |
all | Show environment variables |
-fgw |
all | Disables the embedded graphics |
-g |
all | Run without the GUI or graphics (Linux/UNIX); Run with the GUI minimized (Windows) |
-gr |
all | Run without graphics |
-gu |
all | Run without the GUI but with graphics (Linux/UNIX); Run with the GUI minimized but with graphics (Windows) |
-help |
all | Display command line options |
-hidden |
Windows only | Run in batch mode |
-host_ip=host:ip |
all | Specify the IP interface to be used by the host process |
-i journal |
all | Reads the specified journal file |
-lsf |
Linux/UNIX only | Run FLUENT using LSF |
-mpi= |
all | Specify MPI implementation |
-mpitest |
all | Will launch an MPI program to collect network performance data |
-nm |
all | Do not display mesh after reading |
-pcheck |
Linux/UNIX only | Checks all nodes |
-post |
all | Run the FLUENT post-processing-only executable |
-p |
all | Choose the interconnect |
-r |
all | List all releases installed |
-rx |
all | Specify release number |
-sge |
Linux/UNIX only | Run FLUENT under Sun Grid Engine |
-sge queue |
Linux/UNIX only | Name of the queue for a given computing grid |
-sgeckpt ckpt_obj |
Linux/UNIX only | Set checkpointing object to ckpt_objfor SGE |
-sgepe fluent_pe min_n-max_n |
Linux/UNIX only | Set the parallel environment for SGE to fluent_pe, min_nand max_n are number of min and max nodes requested |
-tx |
all | Specify the number of processors x |
For more information for Fluent text user interface and journal files, please refer to Fluent FAQ.
Submitting Fluent jobs to SLURM
The Fluent simulations can also run in batch. In this section we provide an example script for submitting Fluent jobs to the SLURM scheduler. Please refer to the Running Jobs section of our user guide for detailed tutorials of submitting jobs.
#!/bin/bash
# Job script for submitting a FLUENT job on multiple cores on a single node
# Apply resources via SLURM
#SBATCH --nodes=1
#SBATCH --ntasks=4
#SBATCH --time=01:00:00
#SBATCH --job-name=fluent_test
#SBATCH -o fluent_test_%j.out
#SBATCH -e fluent_test_%j.err
# Loads Ansys and sets the application up
module purge
module load ansys/2022R1
#Initiating Fluent and reading input journal file
fluent 3ddp -t$NTASKS -g -i testJournal.jou
For more information about submitting Fluent jobs, please refer to Fluent FAQ .
Apptainer
Note: Apptainer was formerly known as Singularity and is now a part of the Linux Foundation. When migrating from Singularity see the user compatibility documentation.
Link to section 'What is Apptainer?' of 'Apptainer' What is Apptainer?
Apptainer is an open-source container platform designed to be simple, fast, and secure. It allows the portability and reproducibility of operating systems and application environments through the use of Linux containers. It gives users complete control over their environment.
Apptainer is like Docker but tuned explicitly for HPC clusters. More information is available on the project’s website.
Link to section 'Features' of 'Apptainer' Features
- Run the latest applications on an Ubuntu or Centos userland
- Gain access to the latest developer tools
- Launch MPI programs easily
- Much more
Apptainer’s user guide is available at: apptainer.org/docs/user/main/introduction.html
Link to section 'Example' of 'Apptainer' Example
Here is an example using an Ubuntu 16.04 image on Negishi:
apptainer exec /depot/itap/singularity/ubuntu1604.img cat /etc/lsb-release
DISTRIB_ID=Ubuntu
DISTRIB_RELEASE=16.04
DISTRIB_CODENAME=xenial
DISTRIB_DESCRIPTION="Ubuntu 16.04 LTS"
Here is another example using a Centos 7 image:
apptainer exec /depot/itap/singularity/centos7.img cat /etc/redhat-release
CentOS Linux release 7.3.1611 (Core)
Link to section 'Purdue Cluster Specific Notes' of 'Apptainer' Purdue Cluster Specific Notes
All service providers will integrate Apptainer slightly differently depending on site. The largest customization will be which default files are inserted into your images so that routine services will work.
Services we configure for your images include DNS settings and account information. File systems we overlay into your images are your home directory, scratch, Data Depot, and application file systems.
Here is a list of paths:
- /etc/resolv.conf
- /etc/hosts
- /home/$USER
- /apps
- /scratch
- /depot
This means that within the container environment these paths will be present and the same as outside the container. The /apps, /scratch, and /depot directories will need to exist inside your container to work properly.
Link to section 'Creating Apptainer Images' of 'Apptainer' Creating Apptainer Images
You can build on your system or straight on the cluster (you do not need root privileges to build or run the container).
You can find information and documentation for how to install and use Apptainer on your system:
We have version 1.1.6 (or newer) on the cluster. Please note that installed versions may change throughout cluster life time, so when in doubt, please check exact version with a --version command line flag:
apptainer --version
apptainer version 1.1.6-1
Everything you need on how to build a container is available from their user guide. Below are merely some quick tips for getting your own containers built for Negishi.
You can use a Definition File to both build your container and share its specification with collaborators (for the sake of reproducibility). Here is a simplistic example of such a file:
# FILENAME: Buildfile
Bootstrap: docker
From: ubuntu:18.04
%post
apt-get update && apt-get upgrade -y
mkdir /apps /depot /scratch
To build the image itself:
apptainer build ubuntu-18.04.sif Buildfile
The challenge with this approach however is that it must start from scratch if you decide to change something. In order to create a container image iteratively and interactively, you can use the --sandbox option.
apptainer build --sandbox ubuntu-18.04 docker://ubuntu:18.04
This will not create a flat image file but a directory tree (i.e., a folder), the contents of which are the container's filesystem. In order to get a shell inside the container that allows you to modify it, user the --writable option.
apptainer shell --writable ubuntu-18.04
Apptainer>
You can then proceed to install any libraries, software, etc. within the container. Then to create the final image file, exit the shell and call the build command once more on the sandbox.
apptainer build ubuntu-18.04.sif ubuntu-18.04
Finally, copy the new image to Negishi and run it.
VASP
The Vienna Ab initio Simulation Package (VASP) is a computer program for atomic scale materials modelling, e.g. electronic structure calculations and quantum-mechanical molecular dynamics, from first principles.
Link to section 'VASP License' of 'VASP' VASP License
The VASP team allows only registered users who have purchased their own license to use the software and access is only given to the VASP release which is covered by the license of the respective research group. For those who are interested to use VASP on Negishi , please contact support to request access and provide your email address associated with your license for our verification. Once confirmed, the approved users will be given access to the vasp5 or/and vasp6 unix groups.
Prospective users can use the command below to check their unix groups on the system.
$ id $USER
If you are interested to purchase and get a VASP license, please visit VASP website for more information.
Link to section 'VASP 5 and VASP 6 Installations' of 'VASP' VASP 5 and VASP 6 Installations
Negishi provides VASP 5.4.4.pl2 and VASP 6.4.1 installations and modulefiles with our default environment compiler gcc/12.2.0 and mpi library openmpi/4.1.4. Note that only license-approved users can load the VASP modulefile as below.
You can use the VASP 5.4.4.pl2 module by:
$ module load vasp/5.4.4.pl2
You can use the VASP 6.4.1 module by:
$ module load vasp/6.4.1
Once a VASP module is loaded, you can choose one of the VASP executables to run your code: vasp_std, vasp_gam, and vasp_ncl.
The VASP pseudopotential files are not provided on Negishi, you may need to bring your own POTCAR files.
Link to section 'Build your own VASP 5 and VASP 6' of 'VASP' Build your own VASP 5 and VASP 6
If you would like to use your own VASP on Negishi, please follow the instructions for Installing VASP.6.X.X and Installing VASP.5.X.X.
In the following sections, we provide some instructions about how to install VASP 5 and VASP 6 as well as bash job submit script on Negishi:
VASP Job Submit Script
This shows an example of a job submission file for running VASP pre-built on Negishi:
#!/bin/bash
#SBATCH -A myqueuename # Queue name(use 'slist' command to find queues' name)
#SBATCH --nodes=1 # Total # of nodes
#SBATCH --ntasks=64 # Total # of MPI tasks
#SBATCH --time=1:00:00 # Total run time limit (hh:mm:ss)
#SBATCH -J myjobname # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
# Manage processing environment, load compilers and applications.
module load vasp/5.4.4.pl2 # or module load vasp/6.4.1
module list
# Launch MPI code
mpirun -np $SLURM_NTASKS vasp_std
Build your own VASP 5
For VASP 5.X.X version, VASP provide several templates of makefile.include in the /arch folder, which contain information such as precompiler options, compiler options, and how to link libraries. You can pick up one based on your system and preferred features. Here we provide some examples about how to install the vasp.5.4.4.pl2.tgz version on Negishi with different module environments.
Link to section 'Step 1: Download' of 'Build your own VASP 5' Step 1: Download
As a license holder, you can download the source code of VASP from the VASP Portal, we will not check your license in this case.
Copy the VASP resource file vasp.5.4.4.pl2.tgz to the desired location, and unzip the file tar zxvf vasp.5.4.4.pl2.tgz to obtain the folder /path/to/vasp-build-folder/vasp.5.4.4.pl2and reveal its content.
Link to section 'Step 2: Prepare makefile.include' of 'Build your own VASP 5' Step 2: Prepare makefile.include
We recommend to use GNU compilers parallelized using OpenMPI, combined with MKL for VASP compilation on Negishi.
We are using MKL library, which include BLAS, LAPACK, ScaLAPACK, and FFTW as suggested at VASP wiki, and we can modify makefile.include.linux_gnu from /arch folder:
$ cd /path/to/vasp-build-folder/vasp.5.4.4.pl2
cp arch/makefile.include.linux_gnu makefile.include
Here are the suggested changes for makefile.include, replace the lines between DEBUG=-O0 and OBJECTS= fftmpiw.o fftmpi_map.o fftw3d.o fft3dlib.o with
# Intel MKL (FFTW, BLAS, LAPACK, and scaLAPACK)
LLIBS += -L${MKLROOT}/lib/intel64 -Wl,--no-as-needed -lmkl_gf_lp64 -lmkl_gnu_thread -lmkl_core -lmkl_scalapack_lp64 -lmkl_blacs_openmpi_lp64 -lgomp -lpthread -lm -ldl
INCS = -I$(MKLROOT)/include/fftw
FFLAGS += -march=znver3
# For gcc-10 and higher (comment out for older versions)
FFLAGS += -fallow-argument-mismatch
Remove all the GPU stuff at the end of makefile.include file
Load the required modules:
module --force purge
module load gcc/12.2.0 openmpi/4.1.4
module load intel-mkl/2019.9.304
Link to section 'Step 3: Make' of 'Build your own VASP 5' Step 3: Make
Build VASP with command make all to install all three executables vasp_std, vasp_gam, and vasp_ncl or use make std to install only the vasp_std executable. Use make veryclean to remove the build folder if you would like to start over the installation process.
Link to section 'Step 4: Test' of 'Build your own VASP 5' Step 4: Test
You can open an Interactive session to test the installed VASP with GNU/openMPI compilation, you may bring your own VASP test files:
$ cd /path/to/vasp-test-folder/
module --force purge
module load gcc/12.2.0 openmpi/4.1.4 intel-mkl/2019.9.304
module list
mpirun /path/to/vasp-build-folder/vasp.5.4.4.pl2/bin/vasp_std
Link to section 'Step 5: submit a bash job' of 'Build your own VASP 5' Step 5: submit a bash job
To submit a bash job with your own compiled VASP on Negishi, here is an example about how to set up your environment and launch MPI code.
#!/bin/bash
#SBATCH -A myqueuename # Queue name(use 'slist' command to find queues' name)
#SBATCH --nodes=1 # Total # of nodes
#SBATCH --ntasks=64 # Total # of MPI tasks
#SBATCH --time=1:00:00 # Total run time limit (hh:mm:ss)
#SBATCH -J myjobname # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
# Manage processing environment,load compilers and applications.
module purge
module load gcc/12.2.0 openmpi/4.1.4 intel-mkl/2019.9.304
module list
export PATH=/path/to/vasp-build-folder/vasp.x.x.x/bin:$PATH
# Launch MPI code
mpirun -np $SLURM_NTASKS vasp_std
Build your own VASP 6
For VASP 6.X.X version, VASP provide several templates of makefile.include, which contain information such as precompiler options, compiler options, and how to link libraries. You can pick up one based on your system and preferred features . Here we provide some examples about how to install vasp 6.4.1 on Negishi with different module environments.
Link to section 'Step 1: Download' of 'Build your own VASP 6' Step 1: Download
As a license holder, you can download the source code of VASP from the VASP Portal, we will not check your license in this case.
Copy the VASP resource file vasp.6.4.1.tgz to the desired location, and unzip the file tar zxvf vasp.6.4.1.tgz to obtain the folder /path/to/vasp-build-folder/vasp.6.4.1 and reveal its content.
Link to section 'Step 2: Prepare makefile.include' of 'Build your own VASP 6' Step 2: Prepare makefile.include
We recommend to use GNU compilers parallelized using OpenMPI + OpenMP, combined with MKL for VASP build on Negishi.
We are using MKL library, which include BLAS, LAPACK, ScaLAPACK, and FFTW as suggested at VASP wiki. We can modify makefile.include.gnu_ompi_mkl_omp from /arch folder to fit for system setup:
$ cd /path/to/vasp-build-folder/vasp.6.4.1
$ cp makefile.include.gnu_ompi_mkl_omp makefile.include
Here are the suggested changes for makefile.include:
-
change
VASP_TARGET_CPU ?= -march=nativetoVASP_TARGET_CPU ?= -march=znver3 -
remove
MKLROOT ?= /path/to/your/mkl/installation -
change
LLIBS_MKL =toLLIBS += -
comment out or remove all the lines after
INCS = -I$(MKLROOT)/include/fftw
Then, load the required modules:
$ module purge
$ module load gcc/12.2.0 openmpi/4.1.4
$ module load intel-mkl/2019.9.304
Link to section 'Step 3: Make' of 'Build your own VASP 6' Step 3: Make
Open makefile, make sure the first line is VERSIONS = std gam ncl.
Build VASP with command make all to install all three executables vasp_std, vasp_gam, and vasp_ncl or use make std to install only the vasp_std executable. Use make veryclean to remove the build folder if you would like to start over the installation process.
Link to section 'Step 4: Test' of 'Build your own VASP 6' Step 4: Test
You can open an Interactive session to test the installed VASP 6. Here is an example of testing above installed VASP 6.4.1 with GNU compilers and OpenMPI:
$ cd /path/to/vasp-build-folder/vasp.6.4.1/testsuite
$ module purge
$ module load gcc/12.2.0 openmpi/4.1.4 intel-mkl/2019.9.304
$ ./runtest
Link to section 'Step 5: Submit a bash job' of 'Build your own VASP 6' Step 5: Submit a bash job
To submit a bash job with your own compiled VASP on Negishi, here is an example about how to set up your environment and launch MPI code.
#!/bin/bash
#SBATCH -A myqueuename # Queue name(use 'slist' command to find queues' name)
#SBATCH --nodes=1 # Total # of nodes
#SBATCH --ntasks=64 # Total # of MPI tasks
#SBATCH --time=1:00:00 # Total run time limit (hh:mm:ss)
#SBATCH -J myjobname # Job name
#SBATCH -o myjob.o%j # Name of stdout output file
#SBATCH -e myjob.e%j # Name of stderr error file
# Manage processing environment,load compilers and applications.
module purge
module load gcc/12.2.0 openmpi/4.1.4 intel-mkl/2019.9.304
module list
export PATH=/path/to/vasp-build-folder/vasp.x.x.x/bin:$PATH
# Launch MPI code
mpirun -np $SLURM_NTASKS vasp_std